Carregar apresentação
A apresentação está carregando. Por favor, espere

1
6 de novembro de 2006 Vila Mariana, São Paulo, SP.
I SEMINÁRIO ESTADUAL DE VIGILÂNCIA DAS DOENÇAS PRIÔNICAS NO ESTADO DE SÃO PAULO 6 de novembro de Vila Mariana, São Paulo, SP. Ricardo Nitrini Grupo de Neurologia Cognitiva e do Comportamento do Departamento de Neurologia da FMUSP CEREDIC - Centro de Referência em Distúrbios Cognitivos do Hospital das Clínicas da FMUSP

2
PFG, 49 anos, negra, dona de casa, começou a queixar-se de alterações da visão em dezembro de 2000.
Referia que não distinguia bem os limites dos objetos, que os via com formas e cores diferentes das usuais. Os familiares notaram que a paciente esbarrava em objetos quando andava, principalmente se estivessem situados à sua esquerda.

3
Procurou oftalmologista que não constatou distúrbios de refração e a encaminhou à avaliação neurológica. A avaliação neurológica, em janeiro de 2001, constatou paciente normotensa que não tinha distúrbios de motricidade e sensibilidade mas que não conseguia ler corretamente (as letras pareciam sobrepor-se), embora não tivesse diplopia; tendia a negligenciar o hemicampo visual esquerdo.
, embora não tivesse diplopia; tendia a negligenciar o hemicampo visual esquerdo.")
4
Hemograma, glicemia, uréia, creatinina, transaminases, bilirrubinas, T4 livre e TSH normais.
VHS = 40mm na primeira hora (normal até 20) TC do crânio: normal. EEG: normal LCR: normotenso, límpido e incolor, 1 célula/mm3, 32 mg/dl de proteínas, 87 mg/dl de glicose; reações para sífilis e cisticercose neg.; eletroforese de proteínas: normal.
 TC do crânio: normal. EEG: normal. LCR: normotenso, límpido e incolor, 1 célula/mm3, 32 mg/dl de proteínas, 87 mg/dl de glicose; reações para sífilis e cisticercose neg.; eletroforese de proteínas: normal.")
5
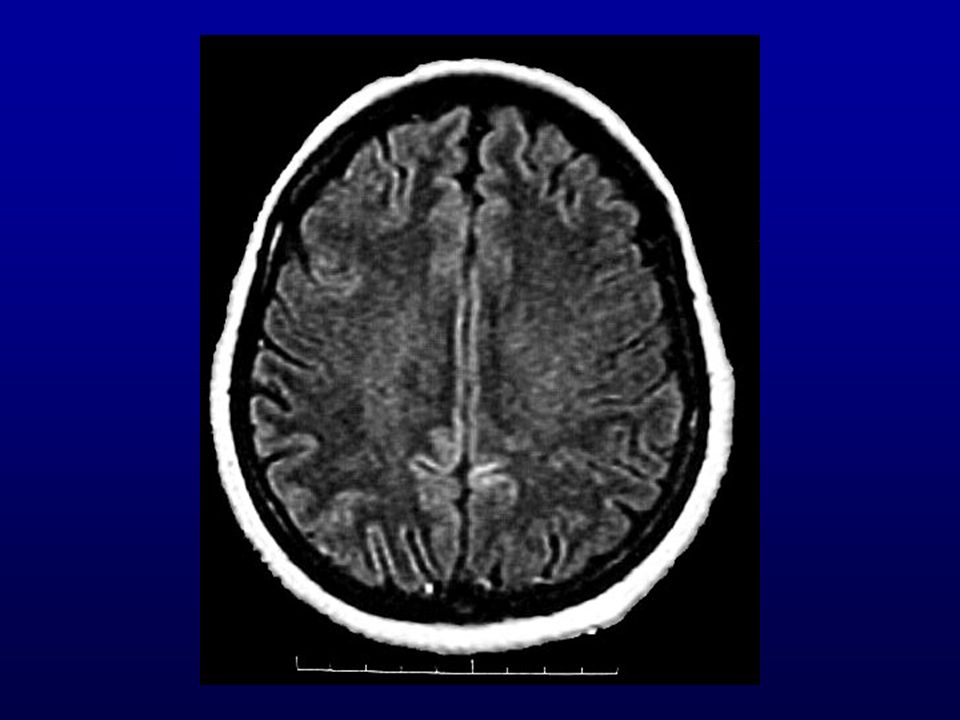
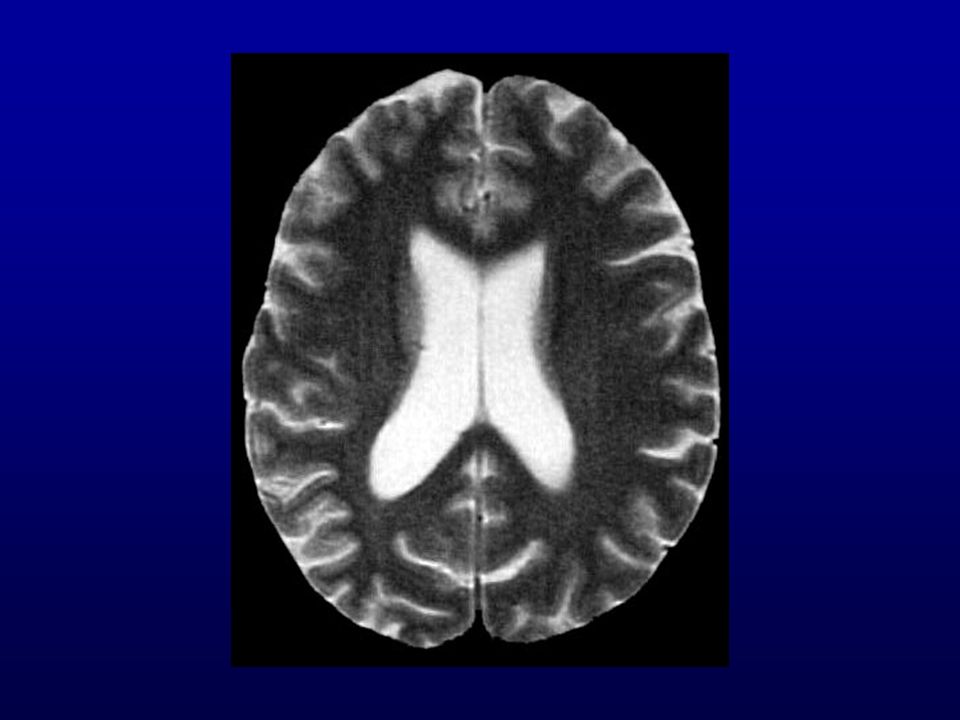
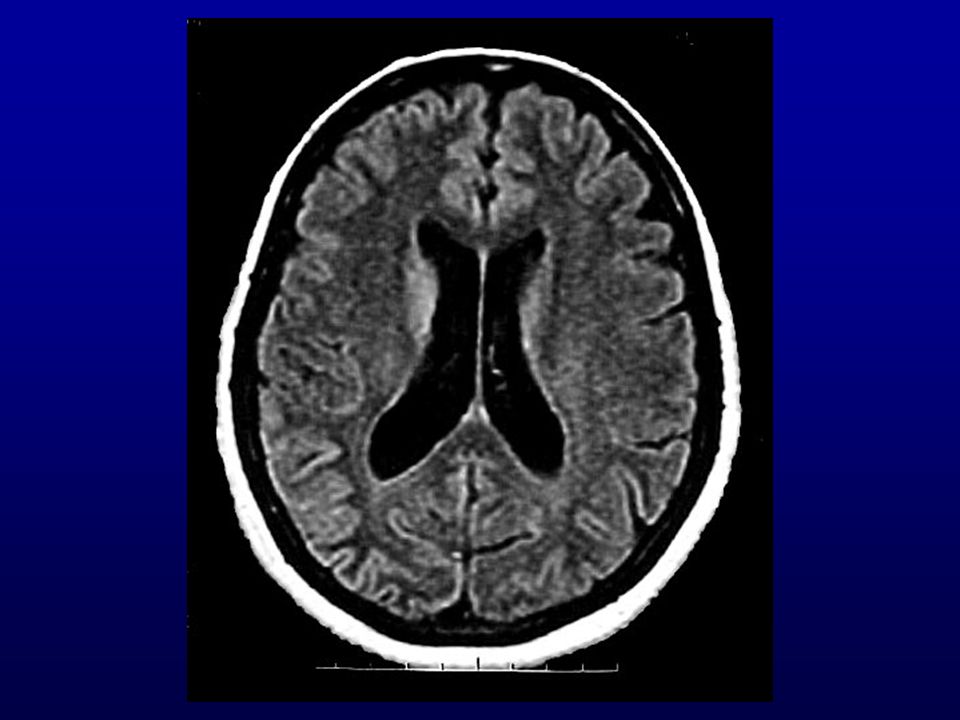
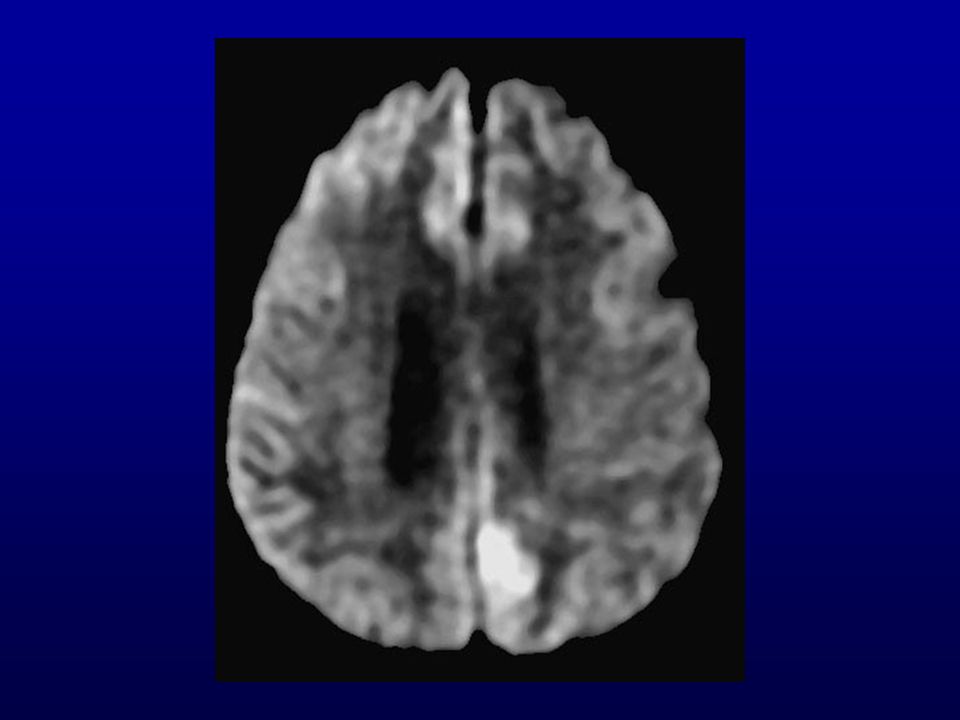
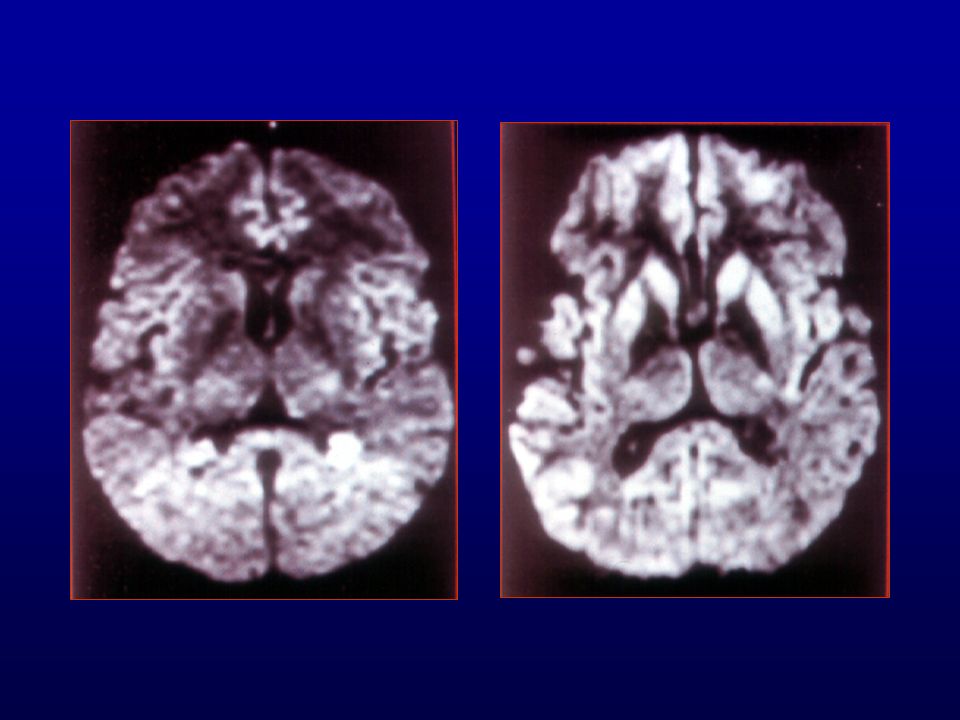
Ressonância magnética (RM) do crânio: lesões isquêmicas (
Ressonância magnética (RM) do crânio: lesões isquêmicas (?) subagudas de limites imprecisos biparieto-occipitais, frontais e dos núcleos caudados e putâmens. Discreta dilatação não-hipertensiva do sistema ventricular supra-tentorial, alargamento dos sulcos e cisternas cerebrais.
 do crânio: lesões isquêmicas ( ) subagudas de limites imprecisos biparieto-occipitais, frontais e dos núcleos caudados e putâmens. Discreta dilatação não-hipertensiva do sistema ventricular supra-tentorial, alargamento dos sulcos e cisternas cerebrais.")
8
Células LE; Anti-DNA nativo; Anti-SSA (ro); Anti SSB (la): dentro dos limites normais.
Fator anti-núcleo: 1/80, pontilhado. Eletroforese de proteínas; proteína C-reativa; prova do látex; anticorpo anticitoplasma de neutrófilos, complemento sérico: dentro dos limites normais. Anticorpos anticardiolipina; anticoagulante lúpico: negativos.

9
Foi medicada com metilprednisolona 1 g/dia IV por dia durante cinco dias, em janeiro de 2001.
Após a alta evoluiu com depressão e embotamento afetivo. Em fevereiro, contatos verbal e visual tornaram-se progressivamente mais pobres, ficando restrita ao leito.

10
Ao exame, foram observados movimentos distônicos dos membros superiores e movimentos involuntários bruscos (abalos), de pequena ou média amplitude nos quatro membros, síncronos ou assíncronos, por vezes de modo maciço, que se repetiam com alta freqüência, de modo arrítmico.
, de pequena ou média amplitude nos quatro membros, síncronos ou assíncronos, por vezes de modo maciço, que se repetiam com alta freqüência, de modo arrítmico.")
11
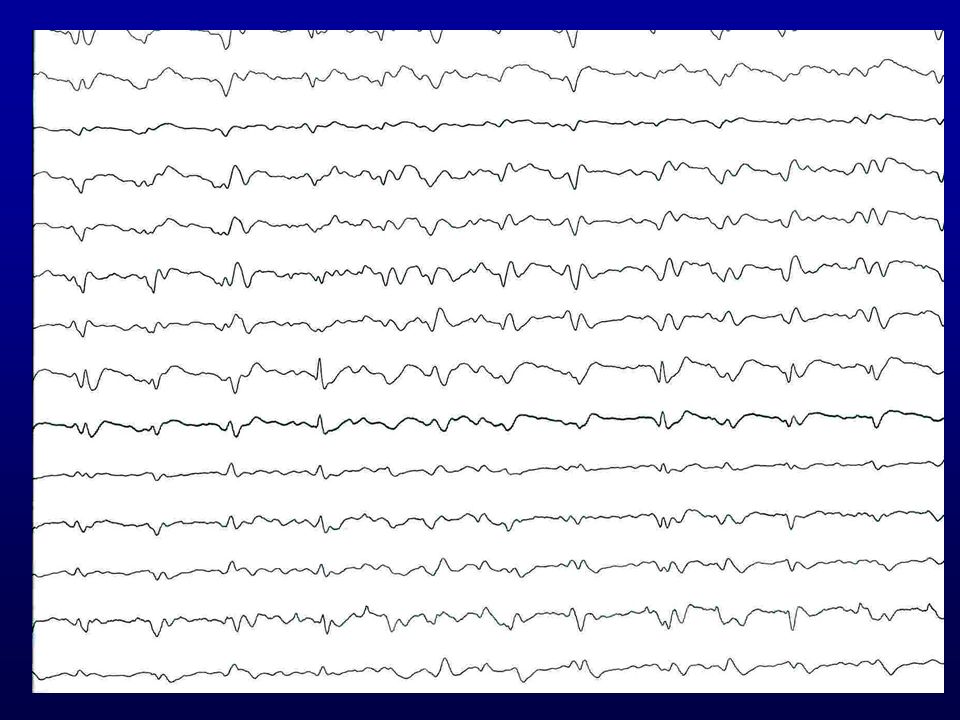
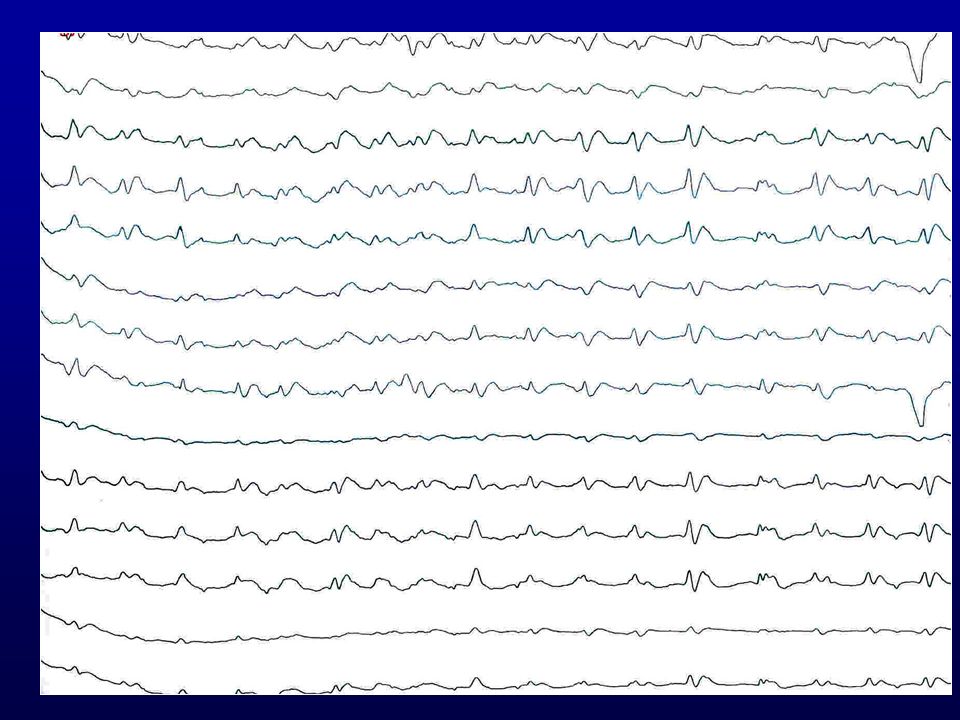
EEG revelou paroxismos de ondas agudas trifásicas repetidos a intervalos regulares de 1 por segundo, aproximadamente, com depressão da atividade de base (atividade periódica curta).
.")
14
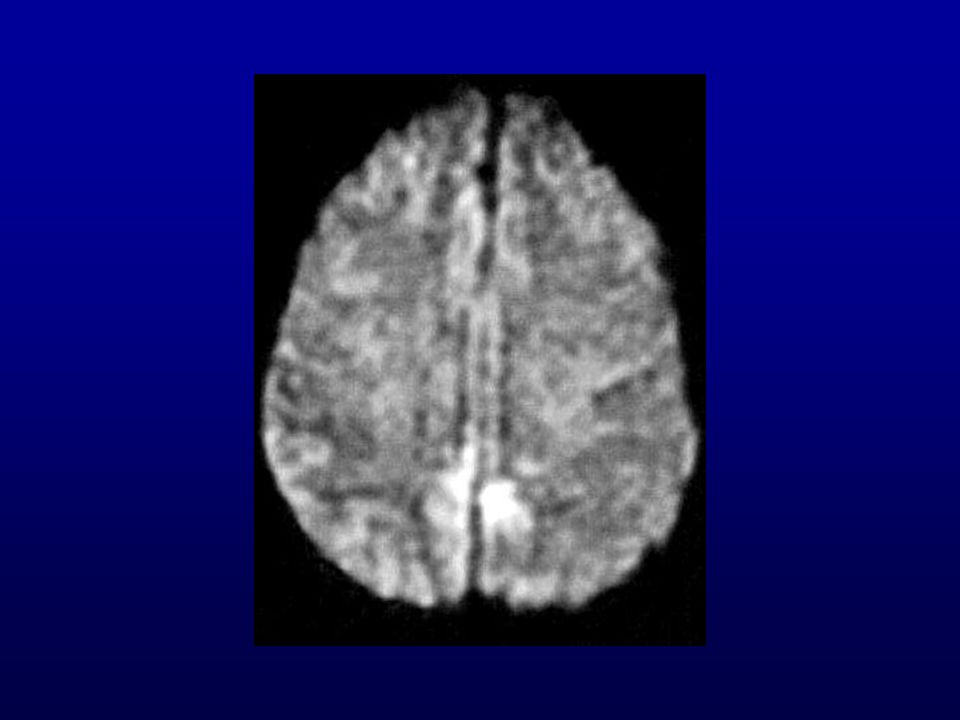
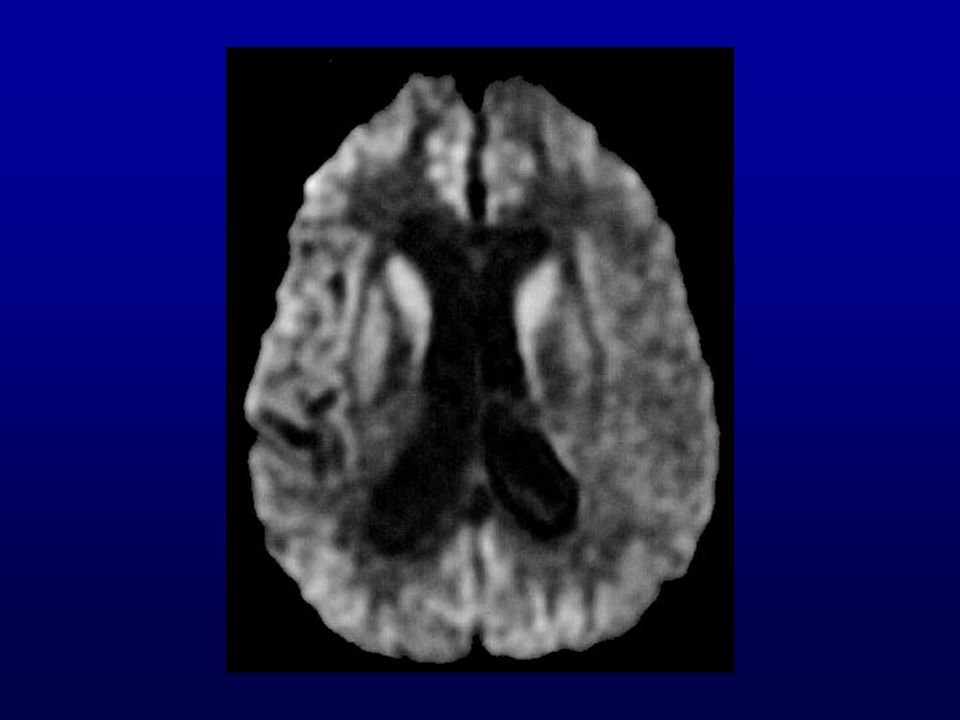
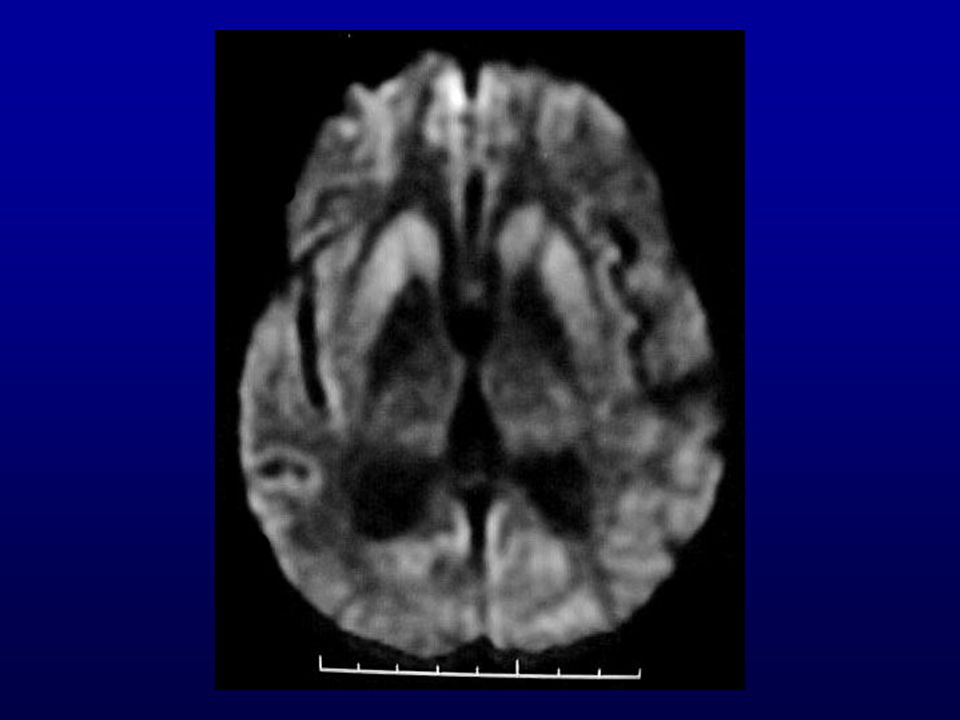
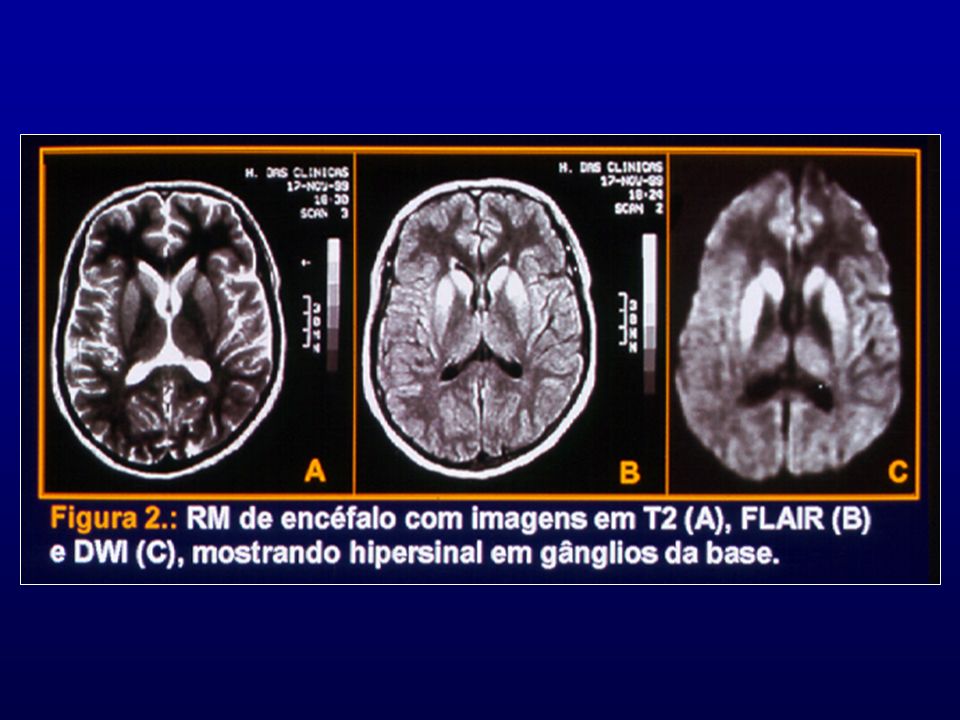
RM: presença de hipersinal em gânglios da base e em áreas corticais mais facilmente visíveis com técnica de difusão.

20
Foi reinternada em abril, quando se encontrava vigil, mas aperceptiva, reagia aos estímulos dolorosos com retirada dos segmentos. (Escore de 8 na escala de Glasgow: 2 pontos em abertura ocular, 4 na melhor resposta motora e 2 na resposta verbal). Mioclonias freqüentes. LCR: presença da proteína Evoluiu com piora progressiva do nível de consciência e faleceu em setembro de 2001, com broncopneumonia, cerca de 10 meses após o início dos sintomas.

21
Anatomia Patológica: DCJ

22
Anatomia Patológica: DCJ

23
CRITÉRIOS DIAGNÓSTICOS DA D. DE CREUTZFELDT-JAKOB
PROVÁVEL: Demência rapidamente progressiva <2anos com atividade periódica no EEG (ou proteína no LCR e pelo menos 2 dos abaixo: mioclonias; sinais cerebelares e/ou visuais; sinais piramidais e/ou extrapiramidais mutismo acinético.

24
CRITÉRIOS DIAGNÓSTICOS DA D. DE CREUTZFELDT-JAKOB
POSSÍVEL: semelhante ao provável mas sem exames complementares sugestivos DEFINITIVO: quadro clínico + encefalopatia espongiforme ou PrPRES no exame neuropatológico

25
Doença de Creutzfeldt-Jakob Esporádica
85 -90% casos de DCJ Igual prevalência entre os sexos Incidência 1 : hab./ ano Idade média de início: 60 anos Duração média: 8 meses

26
Doença de Creutzfeldt-Jakob Esporádica
Quadro Clínico Insônia Astenia Anorexia Pródromos Alt. Comp. e memória Ataxia Nistagmo Diplopia Alucinações Disartria Dist. do Movimento Hiperreflexia Demência global Tremor Sd de Parinaud Sd. Pseudo bulbar Mioclonias Mutismo Incoordenação cerebelar Cegueira Cortical

27
EEG típico de DCJ: Atividade periódica curta

28
ENCEFALOPATIAS SUBAGUDAS COM ATIVIDADE PERIÓDICA NO EEG
P.E.S.A. Alzheimer Men-encefalite herpet. Enc. Hepática Enc. Anóxica Enc. de Hashimoto Enc. Límbica Deg. estriatonigral Gliose subcortical Glioblastoma Metástases Abscessos Neurossífilis Men. Criptocócica Hematoma intracraniano Hiperparatiroidismo, hipercalcemia Li, tricíclicos, Bi

31
Anatomia Patológica: DCJ

32
Doenças Priônicas: Encefalopatias Espongiformes Transmissíveis
Grupo de doenças caracterizadas por: 1)progressiva vacuolização, morte neuronal associada a hipertrofia e proliferação glial 2) presença nos tecidos (principalmente no sistema nervoso) de um tipo de proteína estruturalmente anormal resistente a proteases denominada prion (pronuncia-se príon)
progressiva vacuolização, morte neuronal associada a hipertrofia e proliferação glial. 2) presença nos tecidos (principalmente no sistema nervoso) de um tipo de proteína estruturalmente anormal resistente a proteases denominada prion (pronuncia-se príon)")
33
DOENÇAS PRIÔNICAS Acometem animais e seres humanos Transmissíveis
Material transmissor não é inativado por processos que inativam ácidos nucleicos Podem ser hereditárias (e, simultaneamente, transmissíveis)
")
34
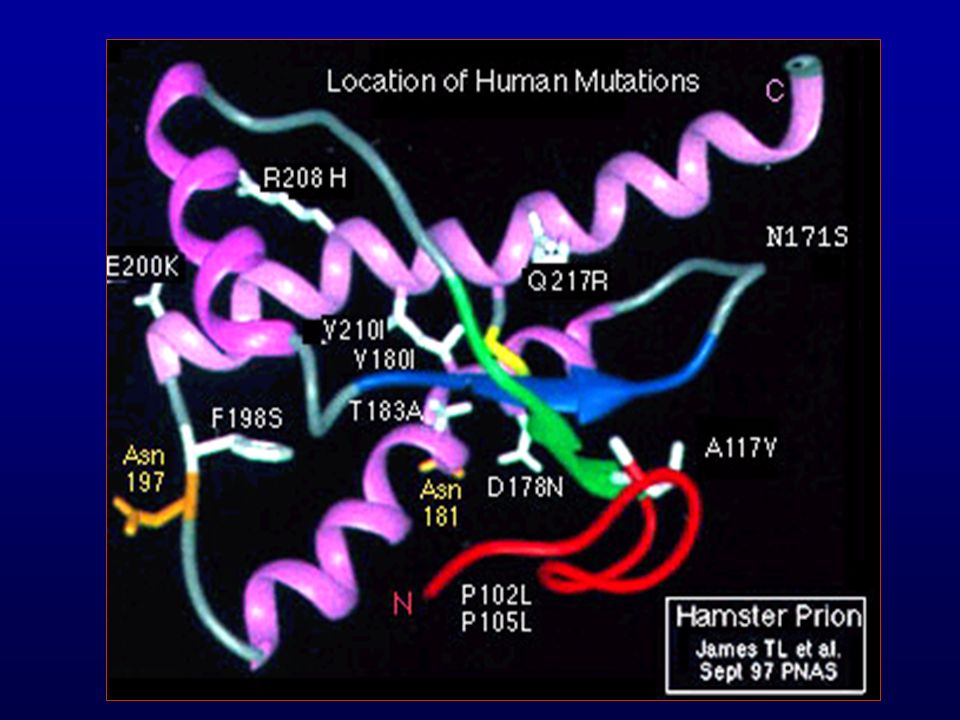
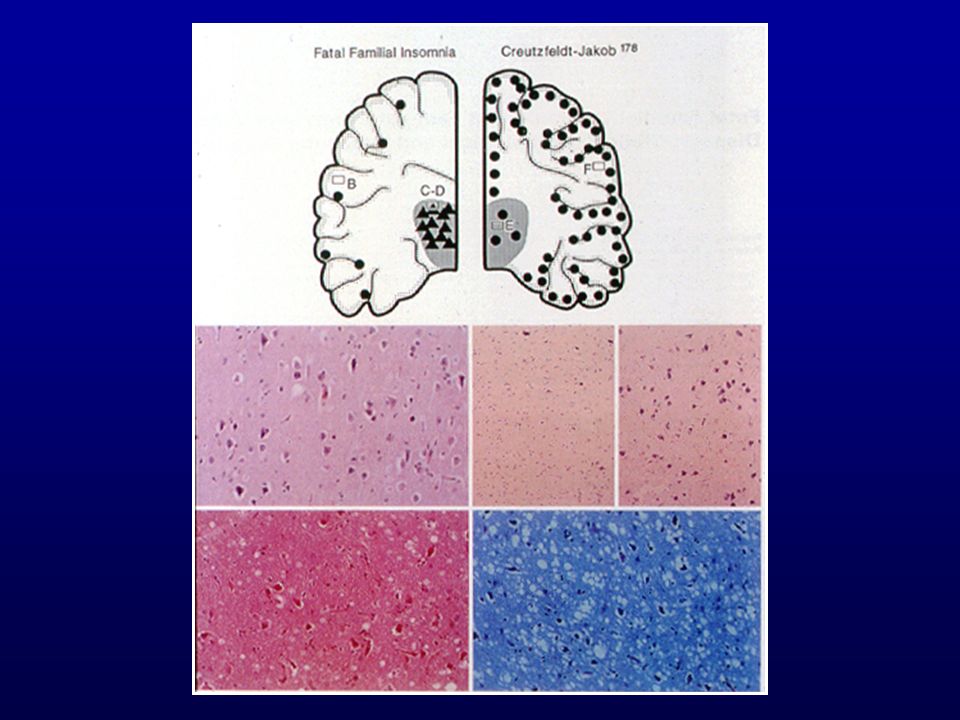
DOENÇAS PRIÔNICAS HUMANAS
Kuru Doença de Creutzfeldt-Jakob Doença de Gerstmann-Sträussler-Scheinker Insônia Fatal Familial

38
D. Carleton Gajdusek (1923- )
Prêmio Nobel 1976

39
Stanley B. Prusiner (1942- ) Prêmio Nobel 1997
 Prêmio Nobel 1997")
40
DOENÇAS PRIÔNICAS A principal hipótese patogênica baseia-se na transformação de uma proteína normal (PrPC) em uma isoforma estruturalmente anormal (PrPSc), parcialmente resistente a proteases. A PrPSc atua como uma fôrma (template), transformando mais unidades de PrPC em PrPSc, que se acumulam no interior da célula, destruindo-a.
 em uma isoforma estruturalmente anormal (PrPSc), parcialmente resistente a proteases. A PrPSc atua como uma fôrma (template), transformando mais unidades de PrPC em PrPSc, que se acumulam no interior da célula, destruindo-a.")
41
DOENÇAS PRIÔNICAS EM ANIMAIS
Scrapie (ovinos) Encefalopatia do mink (Mustela vison) Chronic wasting disease (Cervidae) Encefalopatia espongiforme bovina (BSE) Encefalopatia espongiforme de felinos Encefalopatia espongiforme de ruminantes selvagens em cativeiro
 Encefalopatia do mink (Mustela vison) Chronic wasting disease (Cervidae) Encefalopatia espongiforme bovina (BSE) Encefalopatia espongiforme de felinos. Encefalopatia espongiforme de ruminantes selvagens em cativeiro.")
42
Classificação das Doenças Priônicas Humanas
Esporádicas Hereditárias Adquiridas

43
Doenças Priônicas Humanas Esporádicas
Doença de Creutzfeldt-Jakob Insônia Fatal - Forma esporádica

44
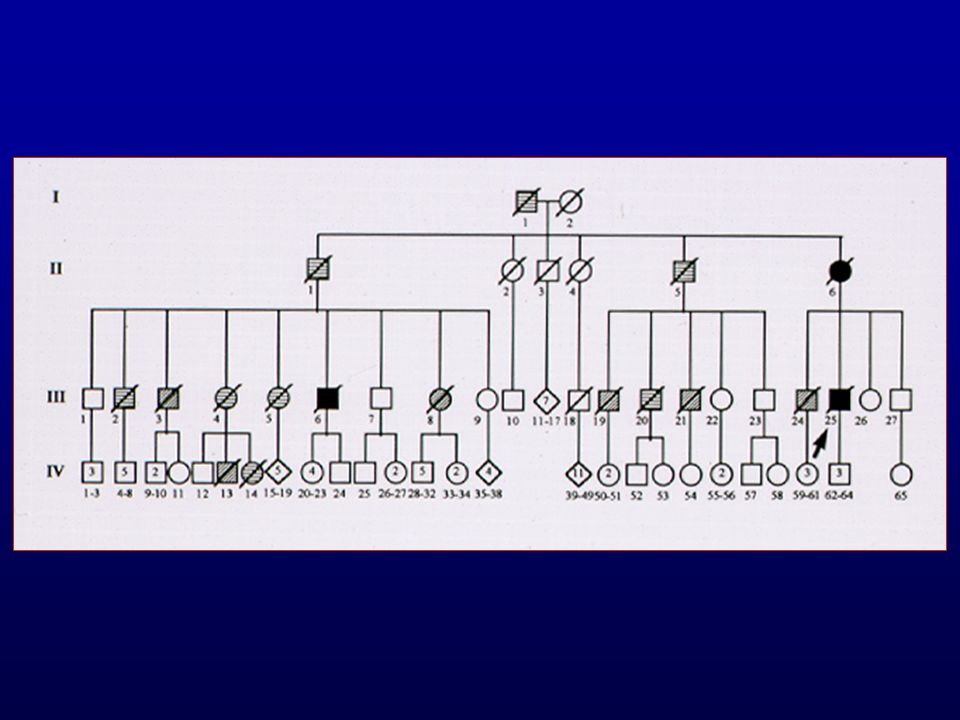
Doenças Priônicas Humanas Hereditárias
D. de Creutzfeldt-Jakob familial Doença de Gerstmann-Sträussler-Scheinker Insônia Fatal Familial Atípicas

45
Doença de Creutzfeldt-Jakob Familiar
% casos Autossômica dominante Início mais precoce Duração mais longa

47
Doença de Creutzfeldt-Jakob Familial
R.Nitrini Codon 183 Início 44 anos Duração de 4 anos Alteração precoce da personalidade Demência fronto-temporal

48
Anatomia Patológica: DCJ (mutação T183A)
")
50
Predominância de homozigose
Polimorfismo no codon 129 Pop. Caucasiana: 37% M/M 51% M/V 12% V/V DCJ esporádico DCJ iatrogênico Predominância de homozigose Está relacionado com a variabilidade do quadro clínico, da evolução e dos achados anatomo-patológicos.

52
Dois subtipos moleculares de proteína priônica
Tipo 1 – cerca de 2/3 dos casos (casos mais típicos) Tipo 2 – menos de 1/3 –exames (14-3-3, EEG, MRI) menos conclusivos ou negativos; clinicamente atípicos; evolução mais longa
 Tipo 2 – menos de 1/3 –exames (14-3-3, EEG, MRI) menos conclusivos ou negativos; clinicamente atípicos; evolução mais longa.")
53
Exames (%) de acordo com subtipo de PrP e polimorfismo no códon 129
EEG TÍPICO MM1 72,8 91,2 MV1 52,9 86,2 VV1 41,7 90,0 MM2 44,4 60,9 MV2 17,5 70,6 VV2 12,8 95,2

54
Doenças Priônicas Humanas Adquiridas
Kuru Doença de Creutzfeldt-Jakob - forma iatrogênica Nova variante da doença de Creutzfeldt-Jakob

55
Doença de Creutzfeldt-Jakob Iatrogênica
Transplante de córnea Hormônio de crescimento de cadáveres Aloenxertos de dura-máter Eletrodos intracorticais

56
NOVA VARIANTE DA DOENÇA DE CREUTZFELDT - JAKOB

57
Nova Variante da DCJ primeiros casos de Encefalopatia espongiforme bovina (BSE) proibido uso de ração com proteína animal; notificação compulsória de BSE

58
Nova Variante da DCJ Centro de controle de unificado para doenças priônicas humanas na GB Casos de DCJ atípicos (nvDCJ) Demonstrado vínculo nvDCJ-BSE

59
Will, R. World Congress of Neurology, Sydney, November 2005.

60
vDCJna Grã-Bretanha (até setembro de 2006) N=156
5 (até 29/09)
 3.")
61
vDCJ fora da Grã-Bretanha (até outubro de 2005)
Outros 10 países: França (9) USA Portugal Itália Holanda Irlanda Espanha Japão Canadá Ar. Saudita
 USA. Portugal Itália. Holanda Irlanda. Espanha Japão. Canadá Ar. Saudita.")
62
Transmissão da vDCJ Não há evidência de transmissão placentária: 9 casos de parto na vigência de vDCJ. Crianças sem a doença, com idades < 10 anos (Will, 2005) Um caso de vDCJ pós-transfusão (Llewelyn et al., Lancet 2004) Outro caso pós-transfusão com PrPSc no baço e linfonodo cervical (sem doença clínica; era heterozigoto no codon 129)
 Um caso de vDCJ pós-transfusão (Llewelyn et al., Lancet 2004) Outro caso pós-transfusão com PrPSc no baço e linfonodo cervical (sem doença clínica; era heterozigoto no codon 129)")
63
Diferenças entre a DCJ esp. e a nvDCJ
ESPORÁDICA NOVA VARIANTE Média de idade de óbito 66 anos 29 anos Mediana da duração 4 meses 13 meses Hipersinal na RM Caudado e putâmen (60%) Tálamo (pulvinar) (90%) no LCR Mais de 90% 50% Imunohistoquím. (Tonsilas) Negativa Positiva EEG Típico (70%) Típico (0%) Will, R. World Congress of Neurology, 2005
 Tálamo (pulvinar) (90%) no LCR. Mais de 90% 50% Imunohistoquím. (Tonsilas) Negativa. Positiva. EEG. Típico (70%) Típico (0%) Will, R. World Congress of Neurology,")
64
Collie et al. AJNR, 2003

65
TRATAMENTO Não há tratamento recomendado para as doenças priônicas. Para as adquiridas, prevenção é essencial Tem sido proposto o emprego de quinacrina e flupirtina para a DCJ mas os efeitos são muito discretos e/ou duvidosos

66
TRATAMENTO Maleato de Flupirtine Quinacrina + Clorpromazina na DCJ
Analgésico não-opióide Efeito citoprotetor in vitro e in vivo sobre neurônios que foram induzidos à apoptose. Quinacrina + Clorpromazina na DCJ

67
CONCLUSÕES Doenças priônicas humanas:
são encefalopatias transmissíveis, geralmente espongiformes podem ser esporádicas, adquiridas ou hereditárias são raras; a mais freqüente é a DCJ

68
CONCLUSÕES Doenças priônicas humanas:
Incluem, além da DCJ, kuru, GSS e IFF. A nova variante da DCJ merece cuidados especiais de acompanhamento epidemiológico Ainda não existe tratamento específico Outras doenças podem ter etiopatogenia similar

Apresentações semelhantes






 se propagam>")




 NO ESTADO DE SÃO PAULO NA DÉCADA DE 90.>")





 NO ESTADO DE SÃO PAULO NA DÉCADA DE 90.>")



