Carregar apresentação
A apresentação está carregando. Por favor, espere

1
BASES BIOQUÍMICAS E MOLECULARES DA DOENÇA GENÉTICA
1. TEORIA UM GENE – UMA ENZIMA UM GENE – UMA PROTEÍNA UM CISTRON – UM POLIPEPTÍDEO MUTAÇÃOPROTEÍNA ALTERADAFUNÇÃO ANORMALDOENÇA - São conhecidas cerca de 500 doenças cujas bases bioquímicas estão determinadas. - Há um grande número de doenças cujas bases bioquímicas não são conhecidas.

2
2. CLASSES FUNCIONAIS DAS PROTEÍNAS E SUAS DOENÇAS (Alguns exemplos):
:")
3
FENILALANINA ----/-- TIROSINA
3. FENILCETONÚRIA 3.1- Quadro Clínico: É variável intra- e extra-familial. É caracterizado por Deficiência Mental, Epilepsia, Pele, Olhos e Cabelos Claros, Sensibilidade da Pele à luz solar com eritema e eczema crônico, Lesão Piramidal, Cheiro característico (urina de rato). 3.2- Bases Metabólicas: FENILALANINA ----/-- TIROSINA Fenilalanina HIDROXILASE Gene localizado em 12q ( Herança = Autossômica Recessiva). Como a fenilalanina não é degradada por pacientes com PKU ela acumula nos fluidos corporais lesando o SNC em desenvolvimento. Uma parte da fenilalanina é transformada em ácidos orgânicos tipo o ácido fenilpirúvico e excretada na urina. O diagnóstico é realizado ou pelo teste do cloreto férrico na urina ou pela dosagem da fenilalanina no sangue.
 Bases Metabólicas: FENILALANINA ----/-- TIROSINA. Fenilalanina HIDROXILASE Gene localizado em 12q ( Herança = Autossômica Recessiva). Como a fenilalanina não é degradada por pacientes com PKU ela acumula nos fluidos corporais lesando o SNC em desenvolvimento. Uma parte da fenilalanina é transformada em ácidos orgânicos tipo o ácido fenilpirúvico e excretada na urina. O diagnóstico é realizado ou pelo teste do cloreto férrico na urina ou pela dosagem da fenilalanina no sangue.")
4
3.3- Triagem Neonatal e Tratamento:
Em virtude de haver tratamento efetivo a triagem neonatal é realizada em muitas regiões do mundo, inclusive algumas do Brasil; pode ser feita pelo teste bacteriológico de Guthrie ou pela dosagem da fenilalanina no sangue (valor normal até 4 mg%). A freqüência é variável, sendo de 1/5000 a 1/60000; no Brasil parece ser de 1/ O teste deve ser realizado com no mínimo dois dias de vida em dieta normal e resultados anormais devem ser investigados para fazer-se o diferencial com as Hiperfenilalaninemias Benignas. O tratamento consiste em restrição dietética durante toda a vida; há leites especiais pobres em fenilalanina.
. A freqüência é variável, sendo de 1/5000 a 1/60000; no Brasil parece ser de 1/ O teste deve ser realizado com no mínimo dois dias de vida em dieta normal e resultados anormais devem ser investigados para fazer-se o diferencial com as Hiperfenilalaninemias Benignas. O tratamento consiste em restrição dietética durante toda a vida; há leites especiais pobres em fenilalanina.")
5
3.4- Genética Molecular da Fenilcetonúria: O Gene da FENILALANINA- HIDROXILASE
Nas populações do Norte da Europa 4 alelos ( 4 mutações ) são responsáveis por 90% dos casos de Fenilcetonúria: 1o) Mutação mais comum ( 38% ) = Mutação do sítio de SPLICE do éxon 12 GT > AT – Forma CLÁSSICA ( GRAVE, atividade enzimática residual abaixo de 1% ) de Fenilcetonúria. 2o) Mutação Missence na Posição 408 da Enzima ( 20% ) = Substituição de Arg..> Trip – Forma Clássica de Fenilcetonúria. 3o) Mutação Missence na Posição 261 da Enzima ( 18% ) = Substituição de Arg > Gln – Forma BENIGNA ( atividade enzimática residual em torno de 5%, compatível com fenótipo normal ) 4o) Mutação Missence na Posição 158 da Enzima ( 14% ) = Substituição de Arg > Gln – Forma Leve de Fenilcetonúria. Nas outras populações, inclusive as do Brasil, há considerável HETEROGENEIDADE GENÉTICA sendo que a maioria dos afetados são COMPOSTOS GENÉTICOS, ou seja, possuem 2 alelos anormais diferentes; isto explica a Heterogeneidade Fenotípica.
 são responsáveis por 90% dos casos de Fenilcetonúria: 1o) Mutação mais comum ( 38% ) = Mutação do sítio de SPLICE do éxon 12 GT > AT – Forma CLÁSSICA ( GRAVE, atividade enzimática residual abaixo de 1% ) de Fenilcetonúria. 2o) Mutação Missence na Posição 408 da Enzima ( 20% ) = Substituição de Arg..> Trip – Forma Clássica de Fenilcetonúria. 3o) Mutação Missence na Posição 261 da Enzima ( 18% ) = Substituição de Arg > Gln – Forma BENIGNA ( atividade enzimática residual em torno de 5%, compatível com fenótipo normal ) 4o) Mutação Missence na Posição 158 da Enzima ( 14% ) = Substituição de Arg > Gln – Forma Leve de Fenilcetonúria. Nas outras populações, inclusive as do Brasil, há considerável HETEROGENEIDADE GENÉTICA sendo que a maioria dos afetados são COMPOSTOS GENÉTICOS, ou seja, possuem 2 alelos anormais diferentes; isto explica a Heterogeneidade Fenotípica.")
6
3.5- DIAGNÓSTICO PRÉ-NATAL
Antes das técnicas da Genética Molecular não era possível o diagnóstico intra-útero da PKU pois a Fenilalanina-Hidroxilase (FAH) não é possível de ser dosada em amniócitos ou vilosidades coriais; hoje é possível este diagnóstico ou pelo estudo dos Haplótipos de RFLP da FAH ou pelo estudo direto das mutações por sequenciamento nas famílias.
 não é possível de ser dosada em amniócitos ou vilosidades coriais; hoje é possível este diagnóstico ou pelo estudo dos Haplótipos de RFLP da FAH ou pelo estudo direto das mutações por sequenciamento nas famílias.")
7
4. FIBROSE CÍSTICA 4.1- Quadro Clínico: É uma doença genética (autossômica recessiva) mais comum em caucasianos = 1/2000 recém-nascidos; heterozigotos = 1/22. Quadro de doença pulmonar crônica, obstrutiva, devido a alteração do muco ( espesso ) e infecções recorrentes; e a Insuficiência Pancreática, devido alteração da secreção das enzimas lipase, tripsina e quimiotripsina, o que leva a quadro de diarréia crônica. Há dois tipos da doença sendo uns SUFICIENTES PANCREÁTICOS ( cerca de 15%) e outros com INSUFICIÊNCIA, sendo o quadro dos primeiros mais leve; estes sub-tipos deve-se a diferentes mutações. Outro tecido atingido é o genital sendo que mais de 95% dos homens afetados que são inférteis, por causa de ausência ou obstrução de vasos deferentes e somente 10% das mulheres que são férteis. O diagnóstico é confirmado pelos níveis elevados de CLORO no suor ( acima de 60 mEq/L); menos de 2% dos pacientes típicos possuem este exame normal.
 mais comum em caucasianos = 1/2000 recém-nascidos; heterozigotos = 1/22. Quadro de doença pulmonar crônica, obstrutiva, devido a alteração do muco ( espesso ) e infecções recorrentes; e a Insuficiência Pancreática, devido alteração da secreção das enzimas lipase, tripsina e quimiotripsina, o que leva a quadro de diarréia crônica. Há dois tipos da doença sendo uns SUFICIENTES PANCREÁTICOS ( cerca de 15%) e outros com INSUFICIÊNCIA, sendo o quadro dos primeiros mais leve; estes sub-tipos deve-se a diferentes mutações. Outro tecido atingido é o genital sendo que mais de 95% dos homens afetados que são inférteis, por causa de ausência ou obstrução de vasos deferentes e somente 10% das mulheres que são férteis. O diagnóstico é confirmado pelos níveis elevados de CLORO no suor ( acima de 60 mEq/L); menos de 2% dos pacientes típicos possuem este exame normal.")
8
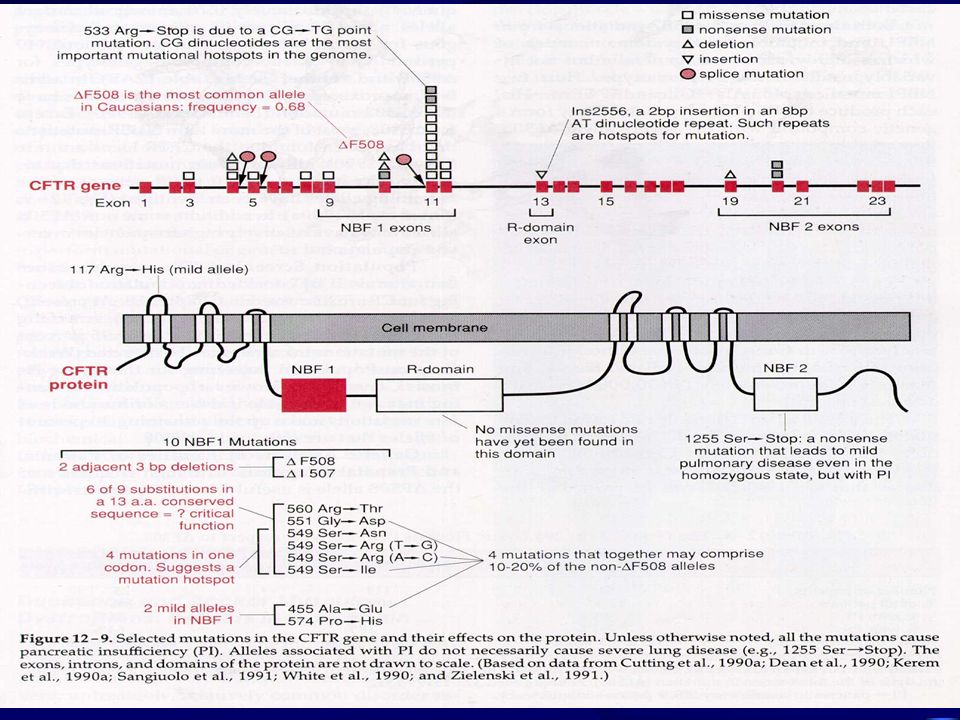
4.2- Bases Metabólicas: O defeito básico é em uma proteína transmembranar de 170 kilodaltons, a qual é chamada de CFTR. Esta proteína forma canais de cloreto e sódio e, regula o transporte destes íons através das membranas das células epiteliais; a alteração desta proteína resulta em desequilíbrios de sal, depletando as células de água e produzindo muco espessado que obstruem os pulmões, o pâncreas, o intestino ( íleo meconial ) e os vasos deferentes e, impede a reabsorção do cloro ao nível das glândulas sudoríparas. Isto leva a fibrose, infiltração de gordura e destruição do parênquima. A proteína CFTR é formada por duas partes principais (NBF 1 e 2), que são os locais de ligação de ATP envolvidos na hidrólise necessária para o transporte de íons (é um canal de cloreto mediado por AMP cíclico) e, que cada um envia um segmento para a membrana que a atravessa 6 vezes; entre estas partes principais fica uma região citoplasmática o domínio R.
 e os vasos deferentes e, impede a reabsorção do cloro ao nível das glândulas sudoríparas. Isto leva a fibrose, infiltração de gordura e destruição do parênquima. A proteína CFTR é formada por duas partes principais (NBF 1 e 2), que são os locais de ligação de ATP envolvidos na hidrólise necessária para o transporte de íons (é um canal de cloreto mediado por AMP cíclico) e, que cada um envia um segmento para a membrana que a atravessa 6 vezes; entre estas partes principais fica uma região citoplasmática o domínio R.")
10
4.3- Genética Molecular da Fibrose Cística:
O gene CF foi mapeado na região 7q31 e clonado; é um gene grande com 250 kb e 27 éxons. A principal, mais freqüente ( cerca de 70% dos alelos) e primeira mutação descrita foi a deleção de três bases que resulta na perda de fenilalanina na posição 508(^F508)localizada no NBF1; hoje são conhecidas mais de 700 mutações, concentradas nos dois domínios de ligação de AT ( o NBF 1 tem cerca de 6 vezes mais mutações que o NBF2); todos os tipos de mutações tem sido descritas, incluindo pequenas deleções , insersões, e mutações de ponto; não foram descritas grandes deleções ou rearranjos. Correlação Genótipo- Fenótipo: A forma mais grave é dos homozigotos ^F508/^F508 (52% entre caucasianos) que apresentam-se com Insuf. Pancreática (I.P.) em 99% dos casos e idade de detecção de 1.8 +/- 3.3 anos; os homozigotos com um alelo ^F508e outro alelo (40%) apresentam-se com I.P. em 72% dos casos e idade de detecção de 4.4 +/- 5.9 anos e, os homozigotos para dois alelos que não o ^F508(8%) apresentam I.P. em somente 36% dos casos e idade de detecção de 8.4+/- 8.3 anos. Em geral há uma razoável correlação entre o genótipo e a função pancreática e uma menor correlação entre o genótipo e a função pulmonar. 4.4- DIAGNÓSTICO PRÉ-NATAL E PRÉ-IMPLANTAÇÃO: É possível ambos através dos estudos do gene CF.
 e primeira mutação descrita foi a deleção de três bases que resulta na perda de fenilalanina na posição 508(^F508)localizada no NBF1; hoje são conhecidas mais de 700 mutações, concentradas nos dois domínios de ligação de AT ( o NBF 1 tem cerca de 6 vezes mais mutações que o NBF2); todos os tipos de mutações tem sido descritas, incluindo pequenas deleções , insersões, e mutações de ponto; não foram descritas grandes deleções ou rearranjos. Correlação Genótipo- Fenótipo: A forma mais grave é dos homozigotos ^F508/^F508 (52% entre caucasianos) que apresentam-se com Insuf. Pancreática (I.P.) em 99% dos casos e idade de detecção de 1.8 +/- 3.3 anos; os homozigotos com um alelo ^F508e outro alelo (40%) apresentam-se com I.P. em 72% dos casos e idade de detecção de 4.4 +/- 5.9 anos e, os homozigotos para dois alelos que não o ^F508(8%) apresentam I.P. em somente 36% dos casos e idade de detecção de 8.4+/- 8.3 anos. Em geral há uma razoável correlação entre o genótipo e a função pancreática e uma menor correlação entre o genótipo e a função pulmonar DIAGNÓSTICO PRÉ-NATAL E PRÉ-IMPLANTAÇÃO: É possível ambos através dos estudos do gene CF.")
11
5. DISTROFIA MUSCULAR PROGRESSIVA (DMP) TIPO DUCHENNE
5.1- Quadro Clínico: Como a herança é ligada ao X recessiva os afetados são os homens; estes são normais até 1o. ou 2 o ano de vida; entre 3 e 5 anos inicia quadro de fraqueza muscular o qual leva a criança a cadeiras de roda entre anos e a morte até os 20 anos( por insuficiência respiratória e/ou cardíaca). Os níveis séricos da enzima muscular CPK (Creatina fosfoquinase) estão muito elevados ( 50 a 100 vezes) desde os estágios pré- clínicos. 5.2- Aspectos Genéticos: Incidência é de 1/3300 recém-nascidos do sexo masculino. A taxa de mutação é calculada em 10 –4, ou seja, considerando-se que um homem produz 80 milhões de espermatozóides por dia, ele produz um gene mutante a cada 10 a 11 segundos. Entre os afetados se considera que i/3 dos casos sejam mutações novas e em 2/3 dos caos a mãe seja portadora (heterozigota). Cerca de 8% das portadoras apresentam alguns sinais clínicos de fraqueza muscular.
. Os níveis séricos da enzima muscular CPK (Creatina fosfoquinase) estão muito elevados ( 50 a 100 vezes) desde os estágios pré- clínicos Aspectos Genéticos: Incidência é de 1/3300 recém-nascidos do sexo masculino. A taxa de mutação é calculada em 10 –4, ou seja, considerando-se que um homem produz 80 milhões de espermatozóides por dia, ele produz um gene mutante a cada 10 a 11 segundos. Entre os afetados se considera que i/3 dos casos sejam mutações novas e em 2/3 dos caos a mãe seja portadora (heterozigota). Cerca de 8% das portadoras apresentam alguns sinais clínicos de fraqueza muscular.")
12
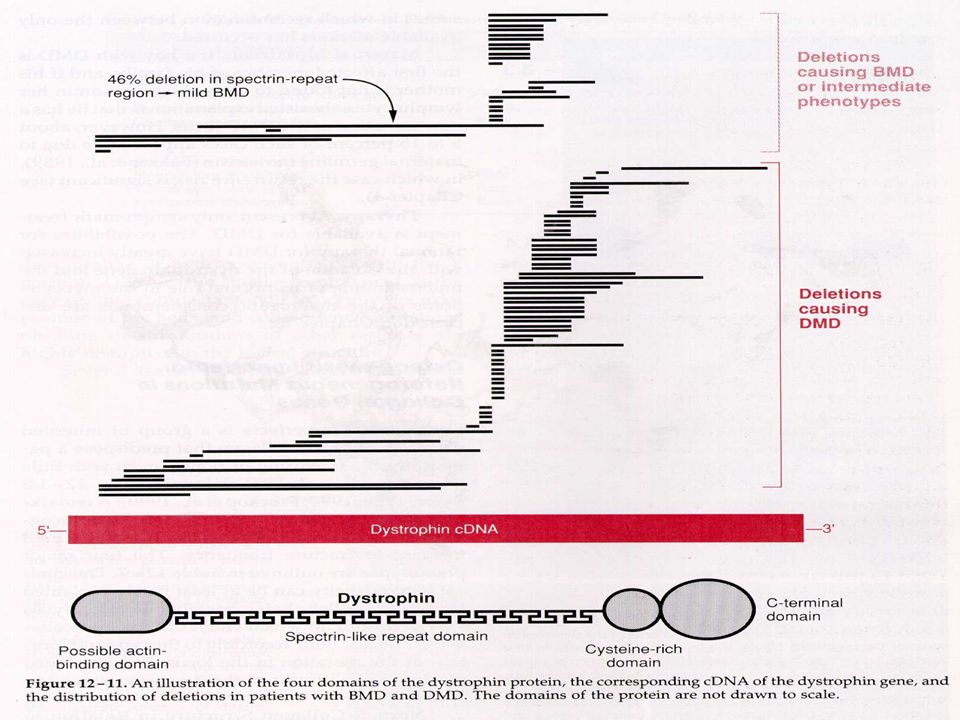
5.3- Genética Molecular da DMPD:
O que mais chama a atenção do gene de DMPD é o seu tamanho – são 2300 kb, representando 1,5% do cromossomo X, com no mínimo 79 éxons e o RNAm de 14 kb, o qual codifica uma proteína chamada DISTROFINA, de 400 kilodalton, com aminoácidos. A distrofina está na parte interna da membrana da célula muscular e é envolvida com a manutenção da integridade da membrana; ela se liga pela parte amino-terminal a F-actina e pela parte carboxi a glicoproteínas importantes para o citoesqueleto (complexo distroglicana-sarcoglicana); na ausência da distrofina a célula muscular degenera .A distrofina é presente nos músculos esqueléticos e cardíacos e no SNC ( numa forma bioquimicamente um pouco diferente). Análise das Mutações da DMDP: Mais d e 60% são deleções de tamanho variável, desde 1 éxon até todo o gene; estas deleções possuem uma distribuição em duas regiões preferenciais, sendo uma parte na metade 5’ do gene e outra parte na parte central do gene. Cerca de 6 a 7% dos pacientes são duplicações parciais do gene e raramente uma síndrome de deleção de genes contíguos. Os pacientes com DMPD não possuem DISTROFINA muscular no músculo.
; na ausência da distrofina a célula muscular degenera .A distrofina é presente nos músculos esqueléticos e cardíacos e no SNC ( numa forma bioquimicamente um pouco diferente). Análise das Mutações da DMDP: Mais d e 60% são deleções de tamanho variável, desde 1 éxon até todo o gene; estas deleções possuem uma distribuição em duas regiões preferenciais, sendo uma parte na metade 5’ do gene e outra parte na parte central do gene. Cerca de 6 a 7% dos pacientes são duplicações parciais do gene e raramente uma síndrome de deleção de genes contíguos. Os pacientes com DMPD não possuem DISTROFINA muscular no músculo.")
14
5.4- DIAGNÓSTICO PRÉ-NATAL E DETECÇÃO DE HETEROZIGOTOS:
Nas famílias em que a mutação é por deleção ou por duplicação é possível o diagnóstico pré-natal através das técnicas de DNA de Southern e PCR. É possível a identificação se uma mulher parente de um afetado é ou não portadora em 75% dos casos usando métodos de DNA e medidas de CPK sérica.

15
6. HIPERCOLESTEROLEMIA FAMILIAL
6.1- Quadro Clínico e Genética Clássica: É o tipo II das HIPERLIPOPROTEINEMIAS, que são caracterizadas por aumento de Lipídeos plasmáticos e das lipoproteínas, no caso aumento do Colesterol e da LDL (lipoproteína de baixa densidade). Estas doenças são muito importantes porque são relacionadas ao Infarto do Miocárdio, É uma doença Autossômica Dominante. Tanto os Homozigotos como os Heterozigotos desenvolvem depósitos de LDL-colesterol na íntima das artérias coronárias (Ateromas), xantomas (depósitos na pele e tendões) e arcus corneae (depósitos na periferia da córnea). Os homozigotos (1/ ) são muito mais afetados com níveis de colesterol que variam de 600 a mg/dl, sendo que a maioria tem infartos antes dos 20 anos, sendo que poucos vivem além dos 30 anos de idade; os heterozigotos (1 em cada 500 indivíduos) apresentam níveis de colesterol entre mg/dl, sendo que cerca de 50% dos homens e 15% das mulheres tem infarto até os 60 anos.
. Estas doenças são muito importantes porque são relacionadas ao Infarto do Miocárdio, É uma doença Autossômica Dominante. Tanto os Homozigotos como os Heterozigotos desenvolvem depósitos de LDL-colesterol na íntima das artérias coronárias (Ateromas), xantomas (depósitos na pele e tendões) e arcus corneae (depósitos na periferia da córnea). Os homozigotos (1/ ) são muito mais afetados com níveis de colesterol que variam de 600 a 1200 mg/dl, sendo que a maioria tem infartos antes dos 20 anos, sendo que poucos vivem além dos 30 anos de idade; os heterozigotos (1 em cada 500 indivíduos) apresentam níveis de colesterol entre mg/dl, sendo que cerca de 50% dos homens e 15% das mulheres tem infarto até os 60 anos.")
16
6.2- Bases Metabólicas: As células normais obtem o colesterol (componente da membrana celular, dos esteróides e dos sais biliares) por síntese de novo ou por captarem do LDL-colesterol exógeno do plasma. O processo de captação é mediado pelo RECEPTOR de LDL( o qual reconhece a apoproteína B-100, a parte protêica do LDL). O receptor de LDL capta o LDL-colesterol e o transfere para dentro da célula por invaginação (endocitose), sendo então levados para os lissosomos aonde são hidrolisados liberando COLESTEROL LIVRE. Este aumento de colesterol livre inibe o processo de síntese endógena tanto de colesterol (pela inibição da enzima HMG Co A redutase) como do receptor de LDL. A doença é devida a redução do número de receptores de LDL na superfície celular, o que diminui a captação do colesterol e leva à seu aumento plasmático.
 por síntese de novo ou por captarem do LDL-colesterol exógeno do plasma. O processo de captação é mediado pelo RECEPTOR de LDL( o qual reconhece a apoproteína B-100, a parte protêica do LDL). O receptor de LDL capta o LDL-colesterol e o transfere para dentro da célula por invaginação (endocitose), sendo então levados para os lissosomos aonde são hidrolisados liberando COLESTEROL LIVRE. Este aumento de colesterol livre inibe o processo de síntese endógena tanto de colesterol (pela inibição da enzima HMG Co A redutase) como do receptor de LDL. A doença é devida a redução do número de receptores de LDL na superfície celular, o que diminui a captação do colesterol e leva à seu aumento plasmático.")
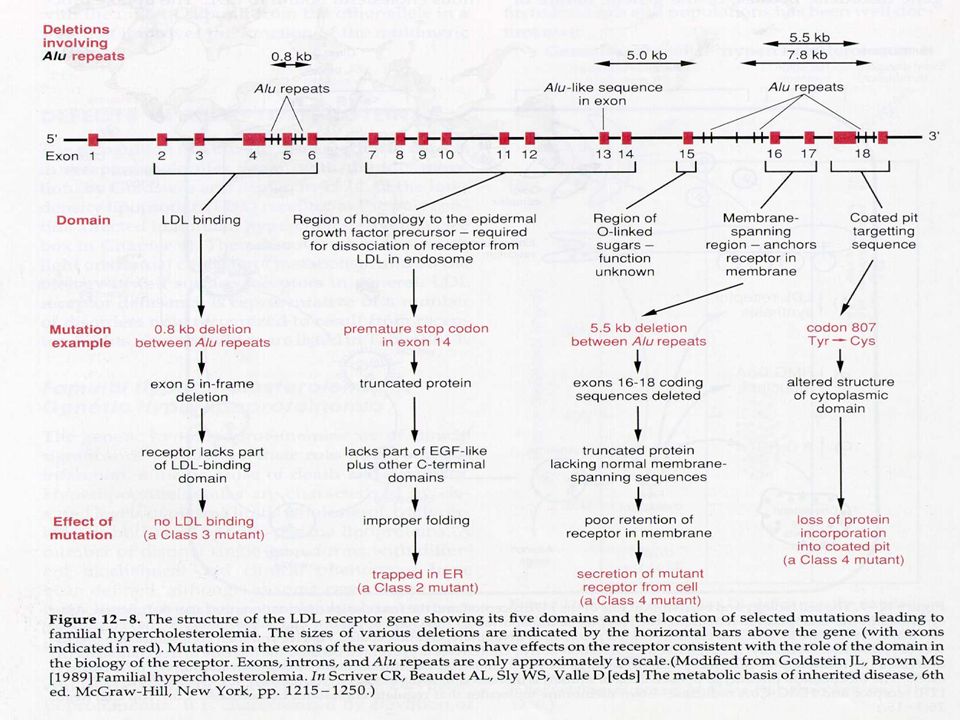
18
6.3- Genética Molecular: O gene foi mapeado no cromosssomo 19, possui 45 kb e consiste de 18 éxons e 17 introns, que codifica um RNAm de 5,3 kb e uma proteína de 839 aminoácidos. Mais de 150 mutações diferentes já foram descritas, sendo mutações de ponto, inserções e deleções. Estas mutações são divididas em 5 classes, dependendo do nível do metabolismo que elas interferem: a) classe I – são as que produzem alelos nulos que não promovem a síntese de nenhuma quantidade de receptor; elas são geralmente deleções. Nas outras classes o receptor é sintetizado, mas tem suas funções alteradas – na classe II o receptor é sintetizado mas não consegue ser transportado do R.E. para o complexo de Golgi e deste para a superfície celular; na classe III o receptor atinge a superfície celular mas são incapazes de ligarem-se com a LDL; na classe IV o receptor é normal mas não se posicionam corretamente na superfície celular e não promovem a internalização do LDL; na classe V o receptor quando entra ligado com o LDL na célula não consegue se desligar não retornado para a superfície celular, sendo degradado.
 classe I – são as que produzem alelos nulos que não promovem a síntese de nenhuma quantidade de receptor; elas são geralmente deleções. Nas outras classes o receptor é sintetizado, mas tem suas funções alteradas – na classe II o receptor é sintetizado mas não consegue ser transportado do R.E. para o complexo de Golgi e deste para a superfície celular; na classe III o receptor atinge a superfície celular mas são incapazes de ligarem-se com a LDL; na classe IV o receptor é normal mas não se posicionam corretamente na superfície celular e não promovem a internalização do LDL; na classe V o receptor quando entra ligado com o LDL na célula não consegue se desligar não retornado para a superfície celular, sendo degradado.")
Apresentações semelhantes