Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Erros inatos do metabolismo
Camila Silva Justo

2
Definição determinadas geneticamente defeito enzimático
bloqueio de uma determinada rota metabólica A conseqüência deste bloqueio é: o acúmulo do substrato , a deficiência do produto da reação, o desvio do substrato para uma rota alternativa O quadro clínico é decorrência destas conseqüências

3
enzima substrato produto enzima produto substrato produto alternativo
NORMAL produto substrato enzima rota alternativa produto alternativo DEFEITO ENZIMÁTICO

4
Quadro clínico consequencia do acúmulo
Depende do tamanho da molécula Pequena se difunde Grande acúmula

5
Características Diminuiu o número de doenças infecciosas
doenças genéticas são 50% dos leitos pediátricos ( Hem. Norte). 10% das doenças genéticas são EIM
. 10% das doenças genéticas são EIM.")
6
Herança Autossômica recessiva Algumas ligadas ao X
Mitocondrias ( 100%) Sozinhas são raras, mas em conjunto têm um incidência de 1:5000. Enzima precisa de 20% para se expressar Recessiva pais hetero 25% Ligado ao X, 50% Incidencia depende da literatura
 Sozinhas são raras, mas em conjunto têm um incidência de 1:5000. Enzima precisa de 20% para se expressar. Recessiva pais hetero 25% Ligado ao X, 50% Incidencia depende da literatura")
7
Homologia fenotípica As características freqüentemente compartilhadas enzimas diferentes funcionam na mesma área de metabolismo doenças diferentes originam-se por defeitos de uma mesma enzima

8
enzimas diferentes funcionam na mesma área de metabolismo
As enzimas 1, 2 , 3 e 4 atuam conjuntamente para gerar o Produto final. Na ausência de qualquer uma destas enzimas o defeito será o mesmo: falta do Produto final Logo a clínica será a mesma! Enzima 1 Enzima 2 Enzima 3 Enzima 4 Produto final

9
doenças diferentes originam-se por defeitos de uma mesma enzima
Produto 1 Produto 2 Produto 3 Produto 4 Enzima Y A enzima Y atua em 4 rotas metabólicas, que produzem produtos diferentes Logo, na ausência da enzima Y todos os 4 produtos estarão ausentes, o que resulta numa mesma clínica

10
Manifestações clínicas
Variadas, desde assintomáticos(glicosúria renal) até fatais ( defeitos no ciclo da uréia) Parecem com doenças comuns (septicemia) Podem aparecer ao nascimento ou na infância Pode ter qualquer sintoma
 até fatais ( defeitos no ciclo da uréia) Parecem com doenças comuns (septicemia) Podem aparecer ao nascimento ou na infância. Pode ter qualquer sintoma.")
11
Achados clínicos precoses em 90 erros metabólicos
N° de doenças Retardo ou regressão neurológicas 31 Hepatomegalia 27 Retardo do crescimento 25 Convulsões 24 Vômitos, letargia 20

12
Diagnóstico Efetivo feito pela determinação da atividade enzimática caro só quando há suspeita Diversidade de efeitos- dificuldade Usa-se testes de triagem (urina, sangue)- falso positivo
- falso positivo.")
13
Teste do pezinho Pelo SUS: fenilcetonúria, hipertiroidismo congênito, homoglobinopatias

14
CLASSIFICAÇÃO

15
Grupos Características Doenças
1- defeito de síntese ou catabolismo de moléculas complexas Sinais e sintomas permanente e progressivos Doença lisossomais e peroxissomias 2- defeito no metabolismo intermediário Intoxicação aguda e crônica;intervalo livre de sintomas; relação com ingestão alimentar Aminoacidopatias; acidurias orgâncias; defeitos do ciclo da uréia e intolerância aos açúcares 3- defeito na produção ou utilização de energia Metabolismo intermediário de fígado, músculo ou cérebro Doenças de depósito de glicogênio; defeitos de ß-oxidação de A.G.; doenças mitocondriasis e hiperlacticemias

16
Grupo I- doenças lisossomais
MUCOPOLISSACARIDOSES ESFINGOLIPIDOSES Hurler Huler- Sacheie Scheie Tay- Sachs Doença de Fabry doença de Gaucher Nieman-Pick

17
MUCOPOLISSACARIDOSES
acúmulo de glicosaminoglicanos leva a desorganização do tecido conjuuntivo(fígado, ossos, córnea) ESFINGOLIPIDOSES Acúmulo de algum lipídio Membranas celulares ( tecido nervoso) clínica
 ESFINGOLIPIDOSES. Acúmulo de algum lipídio. Membranas celulares ( tecido nervoso) clínica.")
18
MPS tipo I ou doença de Hurler

19
Grupo I- mucolipidoses
GLICOPROTEINOSE DISTÚRBIOS DO TRANSPORTE DA MEMBRANA Fucosidose manosidose Sialidose Aspartilglicosaminúrias doença do depósito do ác. Siálico Cistinose Doença de salla

20
Grupo I- mucolipidoses
DOENÇAS DOS PEROXISSOMOS Síndrome de Zellweger Adrenoleucodistrofia (óleo de Lorenzo) Doença de Fersum Hiperoxaluria tipo I
 Doença de Fersum. Hiperoxaluria tipo I.")
21
Grupo II- metabolismo intermediário
Intervalos livres de sintomas Ingesta da alimento nocivo causa crise

22
Grupo II- doenças Cistinúria AMINOACIDOPATIAS Fenilcetonúria
Tirosinemia Homocistinúria Hiperglicemia não-cetótica Doença da urina do xarope de bordo ou leucinose AMINOACIDOPATIAS

23
Grupo II- doenças ACIDÚRIAS ORGÂNICAS Acidemia isovalérica
Deficiência da 3-metilcrotonil CoA carboxilase Acidemia 3- metilglutárica Acidemia proprionica Acidemia metilmalônica Defiencia múltipla da carboxilase Acidemia glutâmica tipo I

24
Acidurias orgânicas Acúmulo de ácidos orgânicos em tecidos e fluídos corpóreos 1:15.000 Acidose metabólica grave, vômitos, letargia, dificuldade de crescimento Correção da acidose, diminuir ingesta de proteínas

25
Grupo II- doenças DEFEITOS DO CICLO DA URÉIA
Deficiencia da carbomoil fosfato sintetase Deficiencia da ornitina transcarbamilase Citrulinemia Acidúria arginosuccínica Argininemia Intolerância lisinúrica á proteína

26
Grupo II- doenças INTOLERÂNCIA AOS AÇÚCARES Galactosemia clássica
Deficiência de galactoquinase Deficiência da epimerase Intolerância hereditária a frutose

27
Aminoacidopatias- fenilcetonuria
Mutações fenilalanina-hidroxilase (converte a fenilalanina em tirosina) A fenilalanina (acúmulo do substrato) é desviada para fenilpiruvato, fenilacetato e fenilactato (produtos alternativos) – tóxicos Causa desmielinização A falta de tirosina (ausência de produto) pertuba a produção de melanina Incidência de 1:5.000 a 1: RN
 A fenilalanina (acúmulo do substrato) é desviada para fenilpiruvato, fenilacetato e fenilactato (produtos alternativos) – tóxicos. Causa desmielinização. A falta de tirosina (ausência de produto) pertuba a produção de melanina. Incidência de 1:5.000 a 1: RN.")
28
fenilcetonuria Proteínas teciduais Proteina da dieta melanina
Proteinas teciduais fenilalanina tirosina tiroxina Fenilalanina hidroxilase Acido fenolpirúvico catecolaminas

29
Hiperfenilalaninemias

30
fenilcetonúria Tratamento: retirada, não total, da fenilalanina
Grávidas que tem FNC, devem ter cuidado rigoroso com a dieta Segundo dia: já ter ingerido proteínas

31
Aminioacidopatias- homocistinúria
cistationina ß- sitase ( enzima da via metabólica da metionina) Incidência: 1: : Diagnóstico: níveis elevados de metionina e homocisteína Quadro clínico: luxação de cristalino em 97% dos pacientes, miopia, osteoporose, escoliose, reatrdo mental, distúrbios do comportamento e fenomenos tromboembólicos.
 Incidência: 1: : Diagnóstico: níveis elevados de metionina e homocisteína. Quadro clínico: luxação de cristalino em 97% dos pacientes, miopia, osteoporose, escoliose, reatrdo mental, distúrbios do comportamento e fenomenos tromboembólicos.")
32
Relações entre FH4, B12 e SAM
responsável pela conversão da homocisteína em cistationina.

33
homocistinúria Tratamento: um grupo responde a piridoxina (B6), outro a betaína e o restante só a dieta pobre em metionina. Todos se beneficiam com o uso do ácido fólico Resposta variável ao tratamento. Até 6 meses de vida diminui risco de complicações
, outro a betaína e o restante só a dieta pobre em metionina. Todos se beneficiam com o uso do ácido fólico. Resposta variável ao tratamento. Até 6 meses de vida diminui risco de complicações.")
34
Doença da urina do xarope de bordo ou leucinose
Enzima deficiente:α-cetoácido desidrogenase de cadeia ramificada Incidencia: 1: Diagnóstico: elevação dos níveis plamáticos de leucina, isoleucina e valina Existem quatro variantes Odor na urina característico de caramelo Evolução rápida, grave cetoacidose Tratamento: medidas de urgência (diálise)
")
35
Metabolismo de aminoacidos ramificados
A alfa-cetoácido desidrogenase de cadeia ramificada é responsável pela descarboxilação oxidativa dos alfa ceto ácidos de aa de cadeia ramificada( valina, leucina, isoleucina) Metabolismo de AA de C.R.
 Metabolismo de AA de C.R.")
36
Defeitos do ciclo da uréia
Via metabólica que metaboliza a amônia pela síntese de arginina e uréia Incidência: 1:30.000 Diagnóstico: hiperamonemia, níveis elavados plasmáticos de glutamina e alanina e a presença de ácido orótico e oritidina na urina Quadro clínico: encefalopatia, alcalose respiratória e hiperamonemia Mais frequente deficiência da transcarbamilase, menos arginase

37
Deficiência das enzimas do Ciclo da Uréia

38
Defeitos do ciclo da uréia
Sintomas têm inicio após 24h de vida: diminuição da aceitação alimentar, letargia que progride para coma Tratamento( aguda): evitar efeitos tóxicos da amônia( hemodiálise, benzoato e fenilacetato) Tratamento:restrição protéica, fórmulas
: evitar efeitos tóxicos da amônia( hemodiálise, benzoato e fenilacetato) Tratamento:restrição protéica, fórmulas.")
39
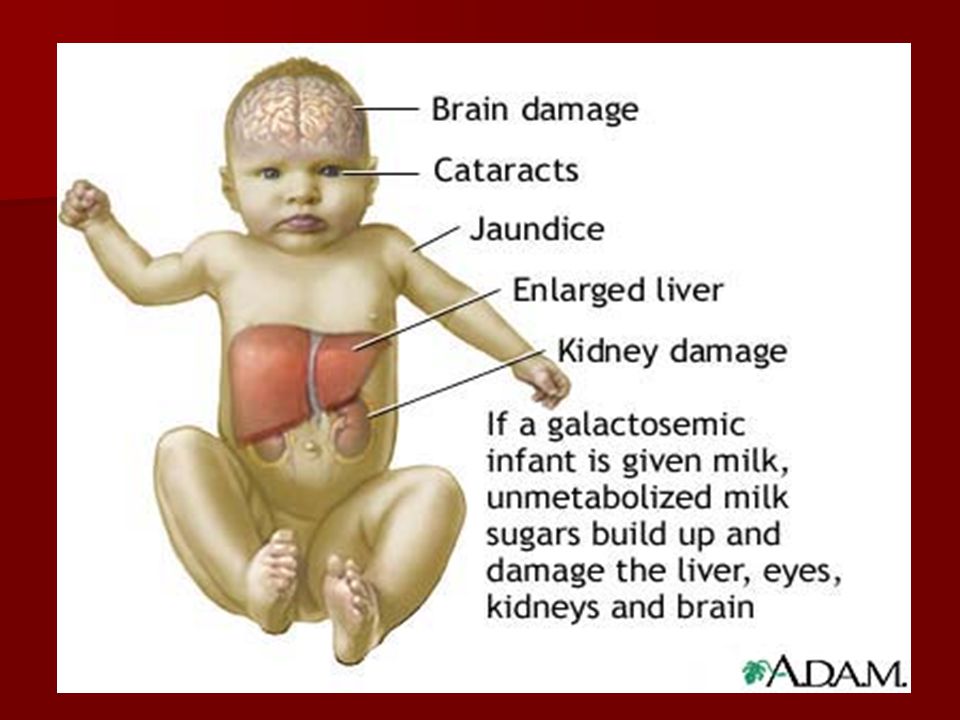
Intolerância aos açúcares- galactosemia clássica

41
Grupo III- defeito na produção ou utilização de energia
Manifestações clínicas: Hipoglicemia Hipotonia Hepatomegalia Miopatia Hiperlacticemia entre outros

42
Grupo III- doenças DEFEITO DE ß-OXIDAÇÃO DE ÁCIDOS GRAXOS Deficiência da Acil-CoA desidrogenase de cadeia média ( MACD) LCAD, SCAD Defeito do transporte plasmático de carnitina Deficiência de carnitina palmitoil transferase

43
Grupo III- doenças DOENÇAS MITOCONDRIAS E HIPERLACTICEMIAS CONGÊNITAS
Defeitos da fosforilação oxidativa Deficiência da carboxiquinase fosfoenolpiruvato Deficiência do comlexo da piruvato desidrogenase Deficiência da piruvato carboxilase Piruvato carboxilase, forma oxalacetato, envolvida na gliconeogênese

44
Grupo III- doenças DOENÇAS DE DEPÓSITO DE GLICOGÊNIO Formas hepáticas
Formas musculares

45
Quando suspeitar de EIM?
SINAIS DE DEGENERAÇÃO DO SNC Desaceleração e parada do desenvolvimento psicomotor Ocorrência de sinais neurológicos anormais (ataxia, espasticidades, convulsões) Progressão de piora inexorável
 Progressão de piora inexorável.")
46
Estratégias de tratamento dos EIM
Restrição alimentar Reposição Desvio

47
Restrição alimentar não ingerir o substrato altamente eficaz
Requer o cumprimento vitalício de uma dieta restrita, artificial e cara Exemplo : hiperfenilalaninemia (PKU),
,")
48
Reposição administrar a enzima ausente
É a forma que tem mais êxito no tratamento Mas existem poucas doenças tratáveis por este mecanismo Exemplo : Doença de Gaucher tipo I e mucopolissacaridoses

49
Desvio Consiste em desviar a rota metabólica alterada para uma rota alternativa desde que o produto da rota alternativa não seja tóxico Exemplo : distúrbios do ciclo da uréia benzoato de sódio+amônia=hipurato.

50
bibliografia Cohn, m. Robert; Roth, Kape . Biochemistry and disease. Ed. Williams e Wilkins, 1996. Martins, Ana Maria. Erros Inatos do metabolismo-abrodagem clínica. Giugliani, Roberto. Erros inatos do metabolismo:uma visão panorâmica Marshall, William J.. Clinical Chemistry. 1995, Ed. Mosby

Apresentações semelhantes













>")





