Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Universidade Estadual de Campinas
Faculdade de Ciências Médicas Departamento de Farmacologia Estudo de Bioequivalência de duas formulações de pantoprazol em voluntários sadios de ambos os sexos Aluna: Maria Carla Petrellis Orientador: Prof. Dr. Gilberto De Nucci 2004

2
Bioequivalência - Considerações
Adoção de uma política de medicamentos genéricos Produção Garantia de Qualidade Prescrição Dispensação e uso É parte fundamental das diretrizes para promoção do uso racional de medicamentos em nosso País

3
Diretrizes da Política de Medicamentos Genéricos
(Brasília ,14 de Abril de 1994) alcançar uma maior racionalidade na utilização de medicamentos estimular a concorrência permitir ao consumidor o acesso de produtos inter- cambiáveis de diferentes preços
 alcançar uma maior racionalidade na utilização. de medicamentos. estimular a concorrência. permitir ao consumidor o acesso de produtos inter- cambiáveis de diferentes preços.")
4
Organização Mundial de Saúde
Glossário de Termos Especializados para Avaliaçâo de Medicamentos (OMS / OPS -1993) - Geneve Um medicamento genérico só deve ter autorizada sua comercialização: Segurança, eficácia e qualidade sejam estabelecidas e documentadas Avaliado sempre em comparação com o medica- mento inovador
 - Geneve. Um medicamento genérico só deve ter autorizada. sua comercialização: Segurança, eficácia e qualidade sejam estabelecidas. e documentadas. Avaliado sempre em comparação com o medica- mento inovador.")
5
Medicamento Genérico: na concepção da OMS,
Definições Medicamento Genérico: na concepção da OMS, “ produto farmacêutico intercambiável” deve apresentar a mesma segurança e eficácia do medicamento de referência produzido após a expiração da proteção patentá- ria ou outros direitos de exclusividade pela DCI (Denominação Comun Internacional), ou seja não apresenta marca.
, ou seja não. apresenta marca.")
6
Medicamento de referência eleito pelo Ministério da Saúde
Deve conter: o mesmo fármaco a mesma dose posológica a mesma forma farmacêutica e via de administração Medicamento de referência eleito pelo Ministério da Saúde

7
Qualidade da formulação e desempenho
PATENTE EXPIRAÇÃO APROVAÇÃO P&D Manufatura Manufatura Adicional EFICÁCIA SEGURANÇA Tempo ESPECIFICAÇÕES Qualidade da formulação e desempenho

8
O medicamento é intercambiável em relação ao medicamento de referência (geralmente o inovador):
possui qualidade comprovada através de teste in vitro e in vivo Sendo considerado seu equivalente terapêutico Ou seja, a mesma eficácia clínica o mesmo potencial para gerar efeitos adversos

9
“Assim, a equivalência terapêutica, é assegurada
através da bioequivalência e constitui a base para a intercambialidade entre o medicamento de referência e o genérico”.

10
Bioequivalência: é um estudo comparativo entre as biodisponibilidades de dois medicamentos que possui: a mesma indicação terapêutica a mesma via de administração na mesma dose

11
Medicamentos Bioequivalentes (F.D.A):
São equivalentes farmacêuticos ou alternativas farmacêuticas que , ao serem administrados: na mesma dose molar, nas mesmas condições experimentais, (seja dose única ou múltipla) Não apresentam diferenças estatísticas significantes quanto: à velocidade de absorção à extensão de absorção
 Não apresentam diferenças estatísticas significantes quanto: à velocidade de absorção. à extensão de absorção.")
12
Parâmetros para determinar a Bioequivalência
ASC (0-t) = Área sob a curva de concentração sangüínea vs tempo Cmax = Pico de concentração plasmática do fármaco t(max) = Tempo para atingir o pico da concentração máxima
 = Área sob a curva de concentração sangüínea. vs tempo. Cmax = Pico de concentração plasmática do fármaco. t(max) = Tempo para atingir o pico da concentração. máxima.")
13
Cmax t max ASC

14
Validação de Métodos Bioanalíticos
Nas últimas décadas Aumento de métodos bioanalíticos aplicados à cromatografia Determinação qualitativa e quantitativa de fár- macos, produtos acabados, matérias primas e amostras biológicas Todas as fases do desenvolvimento do fármaco Pesquisa até a realização de estudos de bioequivalência

15
Entendimento de validação de métodos cromatográficos
Garante nível satisfatório de qualidade de validação metodológica (Massart et al., 1998; Swartz & krull, 1998).
.")
16
Final dos anos 80 Importância da validação de métodos bioanalíticos e suas influências na avaliação e interpretação dos resultados Alvo de amplas discussões presentes em diversas Conferências tanto nos EUA quanto na Europa Fornecer adequadas diretrizes em relação aos parâmetros exigidos na validação analítica Shan et al., 1991 e 1992; Dadgar, 1995; Cartwright et al., 1991 e Arnoux & Morrison, 1992

17
Correta avaliação dos Estudos Biofarmacêuticos
Critérios fundamentais empregados, não só em relação a adequada interpretação desses resultados, como também na aplicação da confiabilidade e na totalidade do desempenho do método bioanalitico. * Pachla et al.,1986.

18
Avaliação da droga e estabilidade do analito
Seletividade/Especificidade Linearidade Precisão e Exatidão Limite de Quantificação Recuperação * Pachla et al., 1986; Buick et al., 1990; MacDouglas e Crummett, 1980; Taylor, 1983;Brooks e Weinfeld,1985; Dadgar e Smith, 1986; Inman et al., 1987 e Shan et al., 1987.

19
Metodologias Cromatografia Líquida de Alta Eficiência (CLAE, LC-MS/MS)
Cromatografia Gasosa (CG, CG-MS/MS) Métodos Imunológicos (RIA, EMIT e ELISA)
 Métodos Imunológicos (RIA, EMIT e ELISA)")
20
na fase de desenvolvimento e também
Métodos analíticos devem ser pré- validados na fase de desenvolvimento e também validados durante o ensaio analítico Garantir que os mesmos sejam satisfatoriamente realizados e que a confirmação da especificações pré-determinadas seja encontrada; Gerar confiabilidade nos resultados obtidos * Buick et al., 1990; Jackson, J. A; 1994; Shan et al., 1992

21
do com as suas propriedades individuais
Os mesmos envolvem procedimentos de determinação e quantificação de moléculas orgânicas com amplas propriedades físico- químicas Cada composto deve ser avaliado de acor- do com as suas propriedades individuais Levando em consideração a complexidade analítica As concentrações almejadas * Jackson, J. A 1994.

22
Seleção e Desenvolvimento
A etapa crítica no Estudo de Bioequivalência é o desenvolvimento e validação de ensaios bioanalíticos Consiste na realização de experimentos Encontrar condições necessárias para a quantificação do analito em questão

23
físico-químicas da droga
Requer conhecimentos das propriedades físico-químicas da droga Processo de desenvolvimento seja racional e individualizado. Todos os critérios envolvidos no desenvolvimento analítico deverão ser registrados em relatórios de acompanhamento analítico *Snyder, 1988; Jackson, J.A 1994

24
Para a aplicação deste fato, consideram-se os
seguintes tipos de validação: Validação Total quando o método é empregado pela primeira vez determinação de um novo analito nesta mesma condição analítica Validação Parcial são modificações do método já validado

25
Protocolo do Ensaio Bioanalítico
Uma vez que o método bioanalítico foi desen- volvido e totalmente validado: manual de procedimentos operacionais padronizados (POPs) para o método deve ser documentado todas as análises do método em questão devem ser submetidas conforme procedimentos do respectivo POP
 para o método deve ser documentado. todas as análises do método em questão devem ser. submetidas conforme procedimentos do respectivo. POP.")
26
As amostras biológicas dos voluntários processadas em duplicata
a serem investigadas processadas em duplicata e compreendidas uma única lista série de listas analíticas

27
Uma Lista analítica é constituída por * :
amostras analíticas em duplicata a ser determinadas. uma curva de calibração em duplicata utilizando a matriz pura, matriz + P.I e 5 á 8 pontos correspon- dendo faixa de aplicação do método. * Shan et al., 1992; Jackson, J.A 1994

28
amostras de controle (n 5) de qualidade de
Baixa concentração (CQA) Média concentração e (CQB) Alta concentração (CQC) dispostos em intervalos para monitorar o desempenho analí- tico.
 Média concentração. e (CQB) Alta concentração (CQC) dispostos em. intervalos para monitorar o desempenho analí- tico.")
29
Parâmetros da Validação Bioanalítica
Especificidade - Seletividade Especificidade: o método produz uma única respos- ta analítica para um único analito

30
Seletividade: a habilidade de uma técnica analítica em distinguir e quantificar, ou não, uma droga em relação a outras resposta de diferentes componentes presentes nos fluidos biológicos

31
emprega-se seis fontes de amostras de matriz biológica branco obtida sob controladas condições, avaliando-se o horário da coleta, ingestão de alimentos, e outros fatores considerados importantes ao método. Cada “branco” deve ser avaliado em relação a interferentes, utilizando-se o método de extração proposto e as condições analíticas em questão (HPLC, LC/MS/MS).
.")
32
Os resultados são confrontados com os obtidos de uma solução aquosa do analíto em concentrações ao Limite de Quantificação (LOQ). Qualquer interferência significativa no tempo de retenção da droga, do metabólitos ou do padrão interno (P.I) deve ser rejeitado Shan et al., 1992; ISO/DIS , 1994
 deve ser rejeitado. Shan et al., 1992; ISO/DIS ,")
33
Interferência em mais que 20% em relação a concentração do LOQ, o método deve ser alterado para eliminar os interferentes Causey et al., 1995; Doing et al., 1990 e Dadgar et al., 1995

34
*International Conference of Harmonization - ICH, Guideline
Linearidade* É considerada como a habilidade de se obter resuldos diretamente proporcionais à concentração do analito na amostra. *International Conference of Harmonization - ICH, Guideline for Validation of Analytical Procedure Methodology, 1996; Comissionof European Communities – Comittee for Proprie- tary Medicinal Products 111/5626/94 final draft, 1994; Buick et al., 1990.

35
A avaliação da curva de calibração, recomendam o uso:
5 a 8 determinações que correspondem ao intervalo entre o valor superior e inferior da substância em exame, que atendam aos requisitos de precisão, exatidão e linearidade. Pennickx et al., 1996; Shan et al., 1992; Hill et al., 1992; Metha, 1898; Massart et al., 1998 e Hartmann et al., 1998.

36
Quatro fatores devem ser empregados na avaliação de curva de calibração:
Precisão e exatidão menor ou igual a 15% nas determinações nominais da curva de calibração, exceto para as concentrações do LOQ. Precisão e exatidão menor ou igual a 20% nas determinações nominais do LOQ.

37
quatro das seis concentrações nominais devem estar de acordo com critérios acima mencionados, incluindo os pontos do LOQ e os pontos de calibração de menor concentração. Valor de coeficiente de correlação linear ou igual ou maior que 0.95.

38
Precisão em uma mesma amostra analítica, em idênticas con-
É definida como grau de concordância entre os resultados de análises individuais, quando o procedimento analítico é aplicado em diversas vezes em uma mesma amostra analítica, em idênticas con- dições experimentais. Swartz e Krull 1998; Buick et al., 1990; Causon, R. 1997; Brittain, 1998; Relatório final da ISO (International Organization for Standardization), 1990/1991; Hartmann et al., 1994; Shan et al., 1992; Buick et al.,1990; USPXX 1990; Brooks, 1985 Buick et al., 1990
, 1990/1991; Hartmann et al., 1994; Shan et al., 1992; Buick et al.,1990; USPXX 1990; Brooks, 1985 Buick et. al.,")
39
Precisão =% CV = (dp/média) x 100
dp = nc2- (c)2 : n (n-1) Onde: CV = coeficiente de variação dp = desvio padrão n = número de dados c= concentração calculada
2 : n (n-1) Onde: CV = coeficiente de variação. dp = desvio padrão. n = número de dados. c= concentração calculada.")
40
Repetibilidade (Precisão intra-ensaio): é a habilida-dade de repetições da metodologia empregada nas mesmas condições laboratoriais, considerando um único dia de análise Reprodutibilidade (Precisão inter-ensaio): é a habilidade de repetições da mesma metodologia aplicada sob diferentes condições laboratoriais, em subseqüentes ocasiões que podem variar semanas ou até meses
: é a habilidade de repetições da mesma metodologia aplicada sob diferentes condições laboratoriais, em subseqüentes ocasiões que podem variar semanas ou até meses.")
41
Exatidão É definida como o grau de concordância
entre os valores individuais encontrados em relação aos valores reais ou nominais * Causon, R. 1997; Swartz e Krull 1998; Buick et al., 1990; Brittain, 1998 e Mehta, 1989.

42
% E.R = (V. exp - V. t) / V. t x 100 Onde,
V. exp: Valores Experimentais V. t: Valores teóricos

43
Normalmente os ensaios para a exatidão
poderão se realizados: Um único dia “ Exatidão Intra-ensaio” Dias diferentes “Exatidão Inter-ensaio”

44
Precisão e Exatidão - Considerações
Emprega-se três níveis de concentração: baixo, que pode ser o LOQ; médio e alto, que estejam dentro do intervalo de limite especificado do procedimento analítico 5 á 10 determinações em replicata para cada nível de concentrações nominais pré - estabelecidas das amostras de controle de qualidade.

45
Limite de Quantificação (LQ)
Representa a mais baixa concentração de um composto de investigação que atende os re- quisitos para precisão e exatidão do ensaio ana- lítico.

46
é estabelecido através da a razão 5:1 entre o sinal e o ruído da linha de base cromatográfica no tempo de retenção do analito de 5 a 6 determinações em replicata para cada nível de concentração pré-estabelecida das amostras de controle de qualidade do LOQ Swartz e Krull, 1998; Causon, R e Buick et al., 1990.

47
Recuperação A recuperação avalia a eficiência da extração
e sua variabilidade. Embora recuperações próximas de 100% sejam desejáveis, baixas recuperações po- dem ser utilizadas (50-60%) se forem precisas, exa- tas e reprodutíveis.
 se forem precisas, exa- tas e reprodutíveis.")
48
Os experimentos de recuperação devem ser
realizados comparando os resuldatos de amostras extraídas a três concentrações diferentes ( alta, média e baixa ) com padrões não extraídos que representam 100%.
 com padrões não extraídos que. representam 100%.")
49
Deve ser considerada durante a fase do desenvolvimento analítico
Estabilidade Deve ser considerada durante a fase do desenvolvimento analítico

50
Curta duração: Congelamento e descongelamento: - 70 ou - 20°C por 3 ciclos; recongelamento de hs; Condições de Análise: °C, tempo máximo de análise do lote; Condições de Auto Sampler: 25°C; tempo máximo de análise do lote;

51
Média duração: temperatura - 20 °C; Avaliar no tempo zero, um, dois quatro, oito e dezesseis dias de armazenamento. Longa duração; deve compreender no tempo entre a data da primeira coleta das amostras e a data da análise da última amostra 3 temperaturas de estocagem, 4, - 20 e - 70ºC

52
Soluções-padrão; Deve ser avaliada à temperatura ambiente por no mínimo 6 horas. Se a solução estoque e padrão interno são refrigeradas ou congeladas por um período de quatorze dias,

53
Um composto é considerado estável se sua decomposição não ultrapassar 20% da sua concentração inicial ou não ultrapassar o limite estabelecido no protocolo de validação

54
Critérios de Aceitabilidade
Precisão: Os CV% do inter-day para os QCs é 15%, e 20% para o LOQ QC, avaliados em um mínimo de 3 lotes. Exatidão: A média dos valores do inter-day devem ser menores que 15% do valor nominal dos QCs, e não desviar mais que 20% para LOQ QC. Sensibilidade: O menor ponto da curva de calibração aceitável como limite de quantificação (LOQ) do método é aquele com CV% 20% (inter-day).
 do método é aquele com CV% 20% (inter-day).")
55
Especificidade: Respostas de picos interferentes no mesmo tempo de retenção do analíto devem ser menores que 20% da resposta de um LOQ. Respostas de picos interferentes no mesmo tempo de retenção do padrão interno (I.S.) devem ser menores que 5% da resposta de um I.S. utilizado nos estudos.
 devem ser menores que 5% da resposta de um I.S. utilizado nos estudos.")
56
Estabilidade: Teste de Longo-Prazo, Curto-Prazo, Freezer and Thaw, Soluções de Estoque e Auto-Sampler são propostos no SOP e variam de estudo para estudo

57
Pantoprazol POTENTE INIBIDOR DE LONGA DURAÇÃO DA BOMBA ÁCIDA DA SUPERFÍCIE DAS CÉLULAS PARIETAIS GÁSTRICAS

59
Objetivos

60
Avaliar a bioequivalência de duas formulações de pantoprazol baseada na comparação dos valores obtidos dos parâmetros farmacocinéticos de cada formulação calculados a partir de medida das concentrações plasmáticas de pantoprazol. Validar a metodologia analítica por LC-MS/MS empregada na quantificação das concentrações de pantoprazol em amostras de plasma humano.

61
Protocolo do Estudo

62
Seleção dos Voluntários
Vinte e dois voluntários sadios de ambos os sexos Idade entre 18 e 45 anos (média= 30 ± 1 ano) Peso Corporal entre 55 e 100 kg (média 77,5 ± 10,1 kg) Entrevista e exames clínicos pré-estudo Informações e esclarecimentos sobre o estudo Assinatura do Termo de Consentimento Livre Esclarecido do estudo Protocolo Clínico Aprovado pela Comissão de Ética da Faculdade de Ciências Médicas da Universidade Federal do Ceará
 Peso Corporal entre 55 e 100 kg (média 77,5 ± 10,1 kg) Entrevista e exames clínicos pré-estudo. Informações e esclarecimentos sobre o estudo. Assinatura do Termo de Consentimento Livre Esclarecido do estudo. Protocolo Clínico Aprovado pela Comissão de Ética da Faculdade de Ciências Médicas da Universidade Federal do Ceará.")
63
Protocolo Clínico Delineamento aleatório cruzado, aberto de dois períodos, com intervalos de 15 dias entre as doses de pantoprazol Hospitalização por 24 horas na Unidade de Farmacologia Clínica Internação seguida de dieta padrão até 23:00 hs Jejum após 23:00hs, e início do estudo clínico às 7:00hs, com administração dose única por via oral de 40 mg de pantoprazol Dieta padrão às 12:00 e às 18:00 hs Restrições: alimentos contendo xantinas

64
Coleta das amostras Amostras de sangue foram coletadas através de escalpe em veia superficial do antebraço do voluntário Os intervalos de coleta: antes (zero), 0.25, 0.5, 0.75, 1.0, 1.25, 1.5, 2.0, 2.5, 3.0, 5.0, 6.0, 8.0 e 12.0 horas Em cada tempo pré-estabelecido de coleta, 10 mL de sangue foram retirados com seringa, através do escalpe, e colocado em tubo limpo pré-heparinizado AS amostras foram centrifugadas durante 10 minutos a 4000rpm Obtenção do soro e armazenamento na temperatura de - 20°C
, 0.25, 0.5, 0.75, 1.0, 1.25, 1.5, 2.0, 2.5, 3.0, 5.0, 6.0, 8.0 e 12.0 horas. Em cada tempo pré-estabelecido de coleta, 10 mL de sangue foram retirados com seringa, através do escalpe, e colocado em tubo limpo pré-heparinizado. AS amostras foram centrifugadas durante 10 minutos a 4000rpm. Obtenção do soro e armazenamento na temperatura de. - 20°C.")
65
Protocolo Analítico Princípio do Método:
Cromatografia Líquida de Alta Eficiência de fase-reversa , acoplada a espectrometria de massa (LCMS/MS) Técnicas auxiliares: Extração Líquido-Líquido com emprego de dietileter:diclorometano (70:30v/v) como solvente extrator
 Técnicas auxiliares: Extração Líquido-Líquido com emprego de dietileter:diclorometano (70:30v/v) como solvente extrator.")
66
Preparo das soluções padrões:
Pantoprazol: a partir de uma solução de uma solução de estoque de pantoprazol (1.0 mg/mL) em metanol:água (50:50) concentração final de 10 g/mL
 em metanol:água (50:50) concentração final de 10 g/mL.")
67
Carbamazepina (P.I): Obs: Todas as soluções padrões foram mantidas a 4°C, sendo substituídas após 2 meses de preparo preparada a partir de uma solução de uma solução de estoque de carbamazepina (1.0 mg/mL) em metanol:água (50:50) concentrações finais de 100 g/mL e 1 g/mL
 em metanol:água. (50:50) concentrações finais de 100 g/mL e 1 g/mL.")
68
Preparo de outras soluções:
Solvente extrator: Dietileter – Diclorometano (70:30) Fase Móvel: 80% Acetonitrila – 0.1% Ácido Trifluoro- acético – 20% Água Deionizada (Sistema Milli-Q)
 Fase Móvel: 80% Acetonitrila – 0.1% Ácido Trifluoro- acético – 20% Água Deionizada (Sistema Milli-Q)")
69
Preparo da curva de calibração e amostras de controle de qualidade (C
Preparo da curva de calibração e amostras de controle de qualidade (C.Q): Curva de Calibração: contaminação de plasma humano controle emprego da solução padrão de pantoprazol (10 g/mL) obtenção dos seguintes pontos da curva de calibração: Branco, Branco + PI, 35, 50, 70, 100, 200, 500 e 1000 ng/mL
: Curva de Calibração: contaminação de plasma humano controle. emprego da solução padrão de pantoprazol (10 g/mL) obtenção dos seguintes pontos da curva de calibração: Branco, Branco + PI, 35, 50, 70, 100, 200, 500 e 1000 ng/mL.")
70
Controle de qualidade:
contaminação de plasma humano controle emprego da solução padrão de pantoprazol (10 g mL) obtenção dos seguintes níveis de concentração: CQA – 120 ng/mL CQB – 800 ng/mL CQC – 600 ng/mL (fator de diluição 1:5) temperatura de armazenamento - 20 °C
 obtenção dos seguintes níveis de concentração: CQA – 120 ng/mL. CQB – 800 ng/mL. CQC – 600 ng/mL (fator de diluição 1:5) temperatura de armazenamento - 20 °C.")
71
Extração de Amostras 0.5 mL de amostras de plasma: voluntários, pontos de da curva de calibração, controle de qualidade em tubos de ensaio 0.05 mL de carbamazepina (1 g/mL), padrão interno adicionado em cada tubo de ensaio breve agitação, repouso por 5 minutos 4 mL de dietileter-diclorometano (70:30), em seguida vigorosa agitação por 30 segundos, seguido centrifu- gação
, padrão interno. adicionado em cada tubo de ensaio. breve agitação, repouso por 5 minutos. 4 mL de dietileter-diclorometano (70:30), em seguida vigorosa agitação por 30 segundos, seguido centrifu- gação.")
72
obtenção da fase orgânica e transferência para tubos de ensaio revestidos de silicone
evaporação em banho- maria (37°C) sob fluxo constante de nitrogênio reconstituição das amostras analíticas com 200 L de fase móvel, em seguida breve agitação acondicionamento das amostras analíticas em vials do injetor automático para posterior análise
 sob fluxo constante de nitrogênio. reconstituição das amostras analíticas com 200 L. de fase móvel, em seguida breve agitação. acondicionamento das amostras analíticas em vials do injetor automático para posterior análise.")
73
Condições Cromatográficas e de Espectrometria de
Massa System Chromatography: consisted of a Hewlett Packard HPLC, binary pump, autosampler injector, degasser, thermostat column, variable-wavelenght UV, coupled a The Quatro II (Micromass, Altringham, Cheshire UK) equipped with a electrospray source Mobile Phase: Acetonitrile:water (40:60) containing 5 mM acetic acid pH= 3.7
 equipped with a electrospray source. Mobile Phase: Acetonitrile:water (40:60) containing 5 mM acetic acid pH= 3.7.")
74
Flow rate: 0.9 mL/min isocratic gradient
Column Chromatography: Genesis C18 4um analytical column (10 mm x 4.0 mm i.d) 40°C Detection: The Quatro II equipped with a electrospray source using a crossflow counter electrode in mode (ES+) and mutiple reaction mode (MRM), monitoring 171.8>127.7 and 306.9>219.8 for metronidazole and fluconazole, repectively. The cone voltage and collission energy were 30V and 20eV, respectively.
 40°C. Detection: The Quatro II equipped with a electrospray source using a crossflow counter electrode in mode (ES+) and mutiple reaction mode (MRM), monitoring 171.8>127.7 and 306.9>219.8 for metronidazole and fluconazole, repectively. The cone voltage and collission energy were 30V and 20eV, respectively.")
75
Resultados

76
Validação Analítica

77
Figura 1 – Cromatogramas de íons (MRM): Plasma Branco (MRM of two channels; ES+ 366.10 91.50)
: Plasma Branco (MRM of two channels; ES 91.50)")
78
Figura 2 – Cromatogramas de íons (MRM): Plasma Lipêmico (MRM of two channels; ES+ 366.10 91.50)
: Plasma Lipêmico (MRM of two channels; ES 91.50)")
79
Figura 3 – Cromatogramas de íons (MRM): Plasma Hemolisado (MRM of two channels; ES+ 366.10 91.50)
: Plasma Hemolisado (MRM of two channels; ES 91.50)")
80
a b Figura 4 – Cromatogramas de íons (MRM): (A) Limite de Quantificação pantoprazol (35ng/mL), (MRM of two channels; ES 91.50). (B) Padrão interno carbamazepina (50ng/mL), (MRM of two channels; ES )
: (A) Limite de Quantificação pantoprazol (35ng/mL), (MRM of two channels; ES 91.50). (B) Padrão interno carbamazepina (50ng/mL), (MRM of two channels; ES )")
81
a b c Figura 5 – Cromatogramas de íons (MRM): (A) Amostra de um voluntário coletada 5 horas após administração da formulação teste (40 mg de panto- prazol), (MRM: 91.50). (B) Plasma contaminado com o padrão interno carbamazepina (50 ng/mL), (MRM: ).(C)Concentração plasmática máxima (99 ng/mL), pantoprazol), (MRM: 91.50) e seu padrão interno, carbamazepina (50ng/mL), (MRM: )
: (A) Amostra de um voluntário coletada 5 horas após administração da formulação teste (40 mg de panto- prazol), (MRM: 91.50). (B) Plasma contaminado com o padrão interno carbamazepina (50 ng/mL), (MRM: ).(C)Concentração plasmática máxima (99 ng/mL), pantoprazol), (MRM: 91.50) e seu padrão interno, carbamazepina (50ng/mL), (MRM: )")
82
a b c Figura 6 – Cromatogramas de íons (MRM): (A) Amostra de um voluntário coletada 3 horas após administração da formulação padrão (40mg de panto- prazol), (MRM: 91.50). (B) Plasma contaminado com o padrão interno carbamazepina (50ng/mL), (MRM: ).(C)Concentração plasmática máxima (145 ng/mL), pantoprazol), (MRM: 91.50) e seu padrão interno, carbamazepina (50ng/mL), (MRM: )
: (A) Amostra de um voluntário coletada 3 horas após administração da formulação padrão (40mg de panto- prazol), (MRM: 91.50). (B) Plasma contaminado com o padrão interno carbamazepina (50ng/mL), (MRM: ).(C)Concentração plasmática máxima (145 ng/mL), pantoprazol), (MRM: 91.50) e seu padrão interno, carbamazepina (50ng/mL), (MRM: )")
83
a b Figura 7 – Espectro de massa: (A) Espectro do pantoprazol adquirido em Q1, energia do cone = 50v. (B) Espectro dos produtos de íons do panto- prazol adquirido em Q2 após colisão induzido dissociação, energia do cone = 50v e energia de colisão – 40eV
 Espectro do pantoprazol adquirido em Q1, energia do cone = 50v. (B) Espectro dos produtos de íons do panto- prazol adquirido em Q2 após colisão induzido dissociação, energia do cone = 50v e energia de colisão – 40eV.")
84
a b Figura 8 - Espectro de massa da carbamazepina: (A) Q1= 30v (B) Q2 = 30v / 25eV
 Q1= 30v (B) Q2 = 30v / 25eV")
85
Figura 9 – Curva de calibração obtida após a contaminação de plasma humano com pantoprazol (35 – 1000 ng/mL) e carbamazepina (50 ng/mL)
 e carbamazepina (50 ng/mL)")
86
Tabela 1- Validação da Curva de Calibração

87
Tabela 2- Validação dos Controles de Qualidade
*Controle de Qualidade 3000ng/mL foi diluído na proporção de 1:5 para quantificação

88
Parâmetros Farmacocinéticos

89
Figura 10- Concentrações plasmáticas (médias aritméticas EP) de pantoprazol em função do tempo para as duas formulações de pantoprazol (40mg) administradas em 22 voluntários sadios de ambos os sexos.
 de pantoprazol em função do tempo para as duas formulações de pantoprazol (40mg) administradas em 22 voluntários sadios de ambos os sexos.")
90
Figura 10- Concentrações plasmáticas (médias aritméticas EP) de pantoprazol em função do tempo para as duas formulações de pantoprazol (40mg) administradas em 22 voluntários sadios de ambos os sexos.
 de pantoprazol em função do tempo para as duas formulações de pantoprazol (40mg) administradas em 22 voluntários sadios de ambos os sexos.")
92
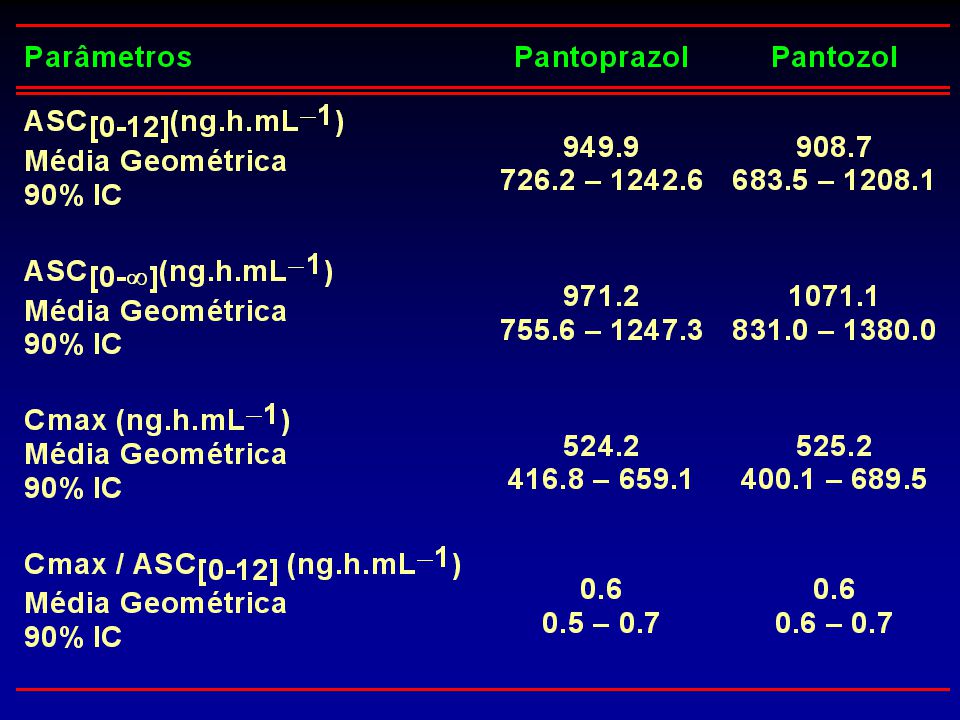
Tabela 3 – Parâmetros farmacocinéticos de pantoprazol obtidos após a administração de cada uma das formulações de pantoprazol (40mg) nos 22 voluntários.
 nos 22 voluntários.")
94
Tabela 4 – Análise estatística das razões individuais teste/referência da ASC 0-12h, ASC0-, Cmax, e das diferenças teste – referência de Tmax entre ambas as formulações de pantoprazol bMédia Aritmética das diferenças individuais c90% das diferenças individuais

95
Conclusão

Apresentações semelhantes


>")







>")










