Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Síndrome de Peutz-Jeghers
Universidade Federal do Estado do Rio de Janeiro Escola de Medicina e Cirurgia Disciplina de Genética II Síndrome de Peutz-Jeghers Igor Gomes Cláudio

2
Introdução A Síndrome de Peutz-Jeghers (SPJ) é uma doença autossômica dominante É caracterizada por polipose hamartomatosa do TGI e pigmentação melânica cutaneomucosa

3
História 1896 John McHutchinson relatou o componente dermatológico em gêmeos idênticos, sendo que um deles morreu de intussuscepção 1921 Foi descrita por Jan Peutz que observou uma relação entre os pólipos intestinais e as máculas mucocutâneas em uma família holandesa 1949 Harold Jeghers descreveu definitivamente a síndrome “Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits” 1954 André J. Bruwer introduziu o epônimo Síndrome de Peutz-Jeghers (SPJ)
")
4
Epidemiologia É uma doença rara Prevalência incerta
Incidência 1/ a 1/ nascidos vivos Não tem predileção por raça ou sexo Média de idade ao diagnóstico 23 anos ♂ e 26 anos ♀

5
Etiologia Caráter hereditário (50%) Mutação no gene STK11 (LKB1)
Padrão autossômico dominante Caráter esporádico (50%) Indivíduos que não têm história familiar Mutação de novo no gene STK11 (?) Outros genes envolvidos (?) MARK
 Indivíduos que não têm história familiar. Mutação de novo no gene STK11 ( ) Outros genes envolvidos ( ) MARK.")
6
O gene STK11 (LKB1) Está localizado no braço curto (p) do cromossomo 19, porção 13.3 (19p13) Codifica a enzima serina/treonina quinase 11
 Está localizado no braço curto (p) do cromossomo 19, porção 13.3 (19p13) Codifica a enzima serina/treonina quinase 11.")
7
A enzima STK11 (LKB1) Supressor tumoral
Ajuda certos tipos de células a se orientar corretamente dentro dos tecidos (polarização) Auxilia na determinação da quantidade de energia que uma célula utiliza Promove apoptose Através de uma combinação desses mecanismos, atua na prevenção de tumores
 Auxilia na determinação da quantidade de energia que uma célula utiliza. Promove apoptose. Através de uma combinação desses mecanismos, atua na prevenção de tumores.")
8
Apresentação Clínica Associação de polipose gastrointestinal e pigmentação mucocutânea Há grande variabilidade de sintomatologia Risco aumentado para desenvolvimento de neoplasias Estômago, cólon e pâncreas Mama, ovário, útero e testículo

9
Polipose gastrointestinal
A idade de aparecimento dos pólipos é variável Mais comum no i. delgado (jejuno > íleo > duodeno) Também pode ocorrer no estômago e no cólon Os pólipos podem causar sangramentos, além de obstrução intestinal recorrente e intussuscepção, exigindo laparotomias repetidas e ressecções intestinais
 Também pode ocorrer no estômago e no cólon. Os pólipos podem causar sangramentos, além de obstrução intestinal recorrente e intussuscepção, exigindo laparotomias repetidas e ressecções intestinais.")
10
Pólipo hamartomatoso Pediculado, avermelhado, brilhante, arredondado, liso, não neoplásico

11
- Uma alça deslizou para dentro de uma outra porção do intestino (intussuscepção) causando inchaço, redução do fluxo sangüíneo, obstrução e dano tecidual. - Exige tratamento de emergência (enema de bário ou cirurgia) para impedir necrose, perfuração intestinal, peritonite e morte.
 para impedir necrose, perfuração intestinal, peritonite e morte..")
12
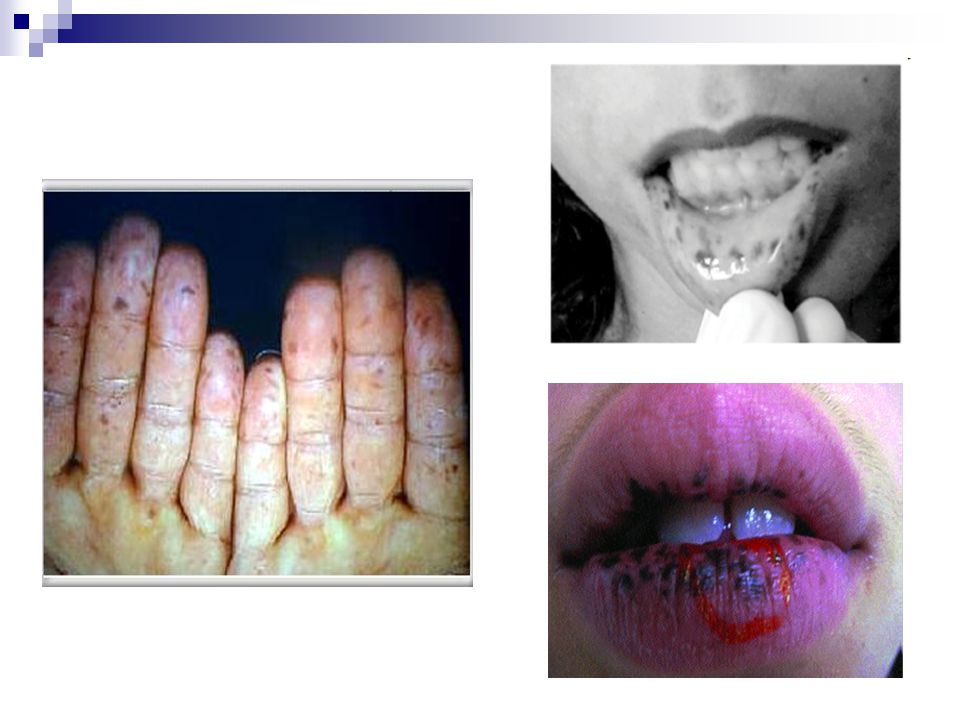
Hiperpigmentação mucocutânea
As máculas surgem na cavidade oral, regiões perioral e periorbitária, dedos das mãos e pés ou genitália Variam em coloração São raramente presentes ao nascimento Tornam-se acentuadas na maioria dos indivíduos antes do quinto ano Podem desaparecer na puberdade e na idade adulta

14
Outras manifestações Episódios repetidos de dor abdominal
Sangramento intestinal inexplicado Prolapso de pólipo retal Irregularidades menstruais e puberdade precoce (devido ao hiperestrogenismo de tumores de cordões sexuais) Ginecomastia e crescimento acelerado (tumores de células de Sertoli) Massa testicular
 Ginecomastia e crescimento acelerado (tumores de células de Sertoli) Massa testicular.")
16
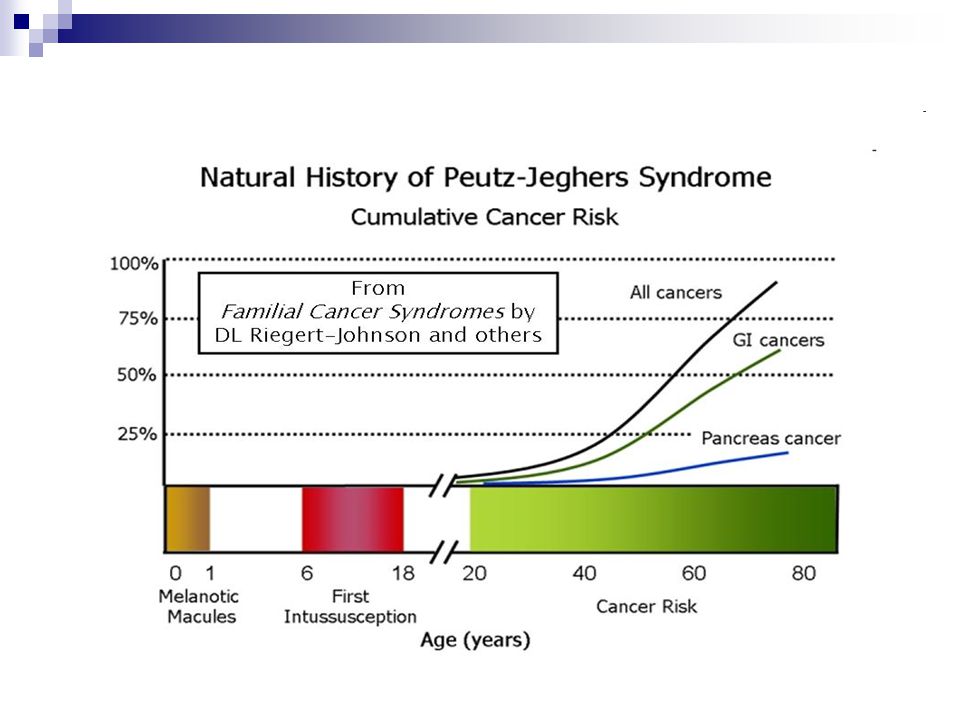
Morbi-mortalidade As manifestações geralmente ocorrem na segunda e terceira décadas de vida Obstrução intestinal e intussuscepção (43%) Dor abdominal (23%), hematoquezia (14%) e prolapso de pólipo colônico (7%) Quase 50% dos pacientes morrem de câncer em torno dos 57 anos de idade A idade média ao primeiro diagnóstico de câncer é 42,9 anos (+ / - 10,2 anos) Noventa por cento dos pacientes apresentam anemia crônica associada à dor abdominal em cólica decorrente de intussuscepções transitórias
, hematoquezia (14%) e prolapso de pólipo colônico (7%) Quase 50% dos pacientes morrem de câncer em torno dos 57 anos de idade. A idade média ao primeiro diagnóstico de câncer é 42,9 anos (+ / - 10,2 anos) Noventa por cento dos pacientes apresentam anemia crônica associada à dor abdominal em cólica decorrente de intussuscepções transitórias.")
17
Diagnóstico Clínico A condição fundamental para o diagnóstico é a presença do pólipo hamartomatoso gastrointestinal Extensa interdigitação da mucosa com feixes de músculo liso Aspecto de pseudoinvasão

18
Diagnóstico Clínico Para os indivíduos com um pólipo hamartomatoso confirmado histopatologicamente, o diagnóstico definitivo da SPJ requer duas das três seguintes condições: História familiar compatível com herança autossômica dominante Hiperpigmentação mucocutânea Pólipos no intestino delgado Para os indivíduos sem comprovação histopatológica dos pólipos hamartomatosos, um diagnóstico provável da SPJ pode ser feito com base na presença de dois dos três critérios anteriores

19
Diagnóstico Clínico Para os indivíduos sem história familiar da SPJ, o diagnóstico depende da presença de dois ou mais pólipos hamartomatosos Para os indivíduos com um parente de primeiro grau com SPJ, a presença de hiperpigmentação mucocutânea é suficiente para o diagnóstico presuntivo Nota: Os indivíduos com SPJ também podem desenvolver outros pólipos, podendo causar confusão com PAF

20
Diagnóstico Laboratorial
Em indivíduos com diagnóstico clínico da SPJ, o teste genético molecular do STK11 (LKB1) detecta mutações no gene da doença 100% dos indivíduos que têm uma história familiar Cerca de 90% naqueles que não têm histórico familiar da SPJ Este tipo de teste está disponível clinicamente 65% das mutações atingem a estrutura da proteína, alterando sua função
 detecta mutações no gene da doença. 100% dos indivíduos que têm uma história familiar. Cerca de 90% naqueles que não têm histórico familiar da SPJ. Este tipo de teste está disponível clinicamente. 65% das mutações atingem a estrutura da proteína, alterando sua função.")
21
Diagnóstico Diferencial
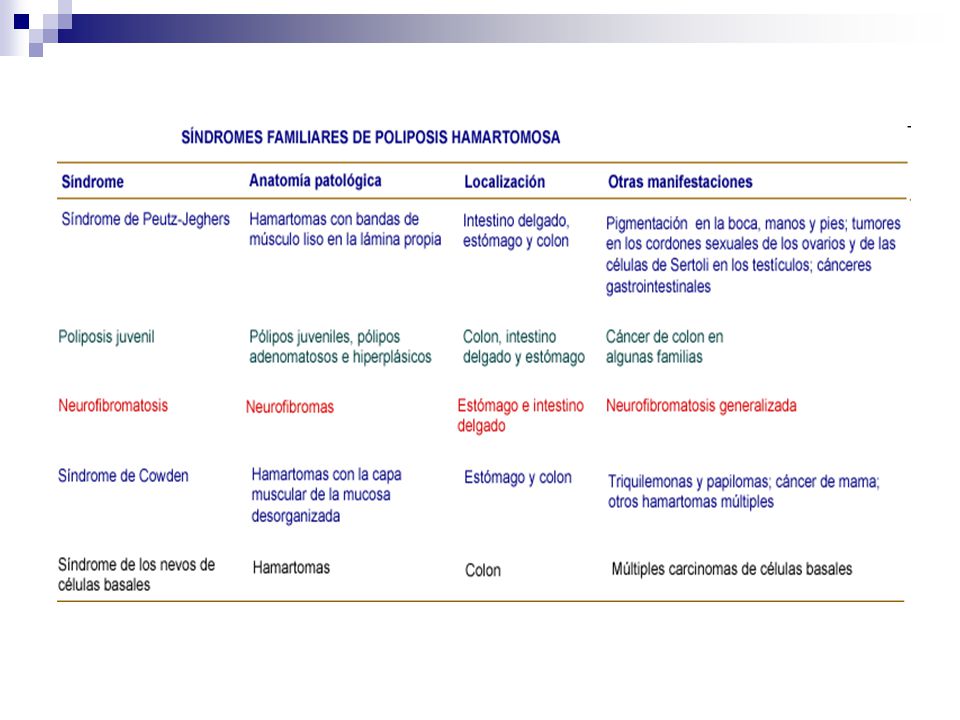
Síndrome Bannayan-Riley-Ruvalcaba Síndrome de Cronkhite-Canadá PAF Síndrome de Gardner Síndrome de polipose juvenil HNPCC Neurofibromatose

23
Tratamento Exame físico anual que inclui a avaliação das mamas, abdome, pelve e os testículos Hemograma completo anualmente Vigilância do câncer: Raio-X de intestino delgado a cada 2 anos EDA e colonoscopia a cada dois anos USG de pâncreas, pelve (mulheres) e testículos (homens) anualmente Mamografia com idades de 25, 30, 35 e 38 anos. Depois, a cada 2 anos até a idade de 50 anos e, em seguida, anualmente Papanicolaou anualmente
 e testículos (homens) anualmente. Mamografia com idades de 25, 30, 35 e 38 anos. Depois, a cada 2 anos até a idade de 50 anos e, em seguida, anualmente. Papanicolaou anualmente.")
24
Tratamento - Pólipos O tratamento cirúrgico está indicado principalmente nos casos de obstrução, sangramento e intussuscepção A tendência é que estes procedimentos sejam conservadores (polipectomias ou ressecções segmentares) Como os pólipos se distribuem por todo o trato digestivo, qualquer tentativa de ressecções mais extensas não terá êxito no controle da doença, além de piorar o estado nutricional desses pacientes O método endoscópico, além de permitir o diagnóstico e localização das lesões, tem um papel importante na terapêutica possibilitando a ressecção de pólipos isoladamente Restringir enterectomia aos segmentos com complicações
 Como os pólipos se distribuem por todo o trato digestivo, qualquer tentativa de ressecções mais extensas não terá êxito no controle da doença, além de piorar o estado nutricional desses pacientes. O método endoscópico, além de permitir o diagnóstico e localização das lesões, tem um papel importante na terapêutica possibilitando a ressecção de pólipos isoladamente. Restringir enterectomia aos segmentos com complicações.")
25
Aconselhamento Genético
Herança autossômica dominante Cerca de 50% dos probandos têm um dos pais afetado e cerca de 50% não têm história familiar da SPJ A proporção de casos causados por mutações de novo é desconhecida, pois a freqüência dos sinais sutis da doença nos pais não tem sido devidamente avaliada Dados moleculares são insuficientes

26
Aconselhamento Genético
O risco para a prole de um probando com história familiar é de 50% O risco para a prole de um probando sem história familiar é de 50%, se for identificada uma mutação no gene STK11 Caso não seja detectada essa alteração, o risco permanece desconhecido Os pais dos indivíduos afetados, sem história familiar conhecida da SP, devem ser avaliados clinicamente e com testes de genética molecular, se uma mutação STK11 foi identificada no probando Até o momento, não existem relatos de testes de pré-natal para a SPJ, embora seja tecnicamente possível, em famílias com mutações conhecidas

27
Referências Bibliográficas

28
Obrigado!!!

Apresentações semelhantes