Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Química Orgânica

2
C C C + Y Z Y + Z Y Z d+ d- Química da ligação p + -
electrófilo nucleófilo C C Y + d+ d- Y Z + Reacção típica dos alcenos é a adição Todas as reacções são iniciadas com um ataque a uma espécie electrofílica e consquente ligação deste a um dos carbonos sp2 + Z - Esta descrição, passo a reagentes são transformados em produtos tem a designação de mecanismo da reacção passo, da forma como os C Y Z Em seguida ocorre a adição da espécie nucleofílica ao outro carbono sp2 As reacções de adição a alcenos ocorrem porque há libertação de energia Adição electrofílica

3
H—X C C HF HCl HBr HI << < < H X –
Adição electrofílica C H—X C H X – H—X HF HCl HBr HI Reatividade de HX << < < X = Cl, Br, I A ordem de reactividade é consequência da capacidade do ácido em doar protões

4
Adição electrofílica de HX
Mecanismo: A reacção processa-se tendo como passo limitante a formação de um carbocatião como intermediário C X H .. : .. : X – C H + Ataque electrofílico do par de electrões p ao protão do HX H C X O carbocatião gerado reage com o ião haleto, formando o haleto de alquilo correspondente

5
Adição electrofílica de HX
Qual é o produto da reacção? ou A formação do carbocatião é o passo limitante Só se forma este produto + Então o carbocatião que lhe dá origem é o mais estável dos dois possíveis

6
Estabilidade de carbocatiões
Carbocatiões são espécies de alta energia, instáveis e consequentemente muito reactivas Não são isolados, mas podem ser detectados por métodos espectroscópicos Estabilidade de carbocatiões catião metilo carbocatião primário carbocatião secundário carbocatião terciário Estabilidade crescente A designação dos carbocatiões é dada de acordo com o tipo de carbono que apresenta a carga positiva Maior número de grupos alquilo ligados ao carbono carregado positivamente, implicam maior estabilidade do carbocatião

7
Dispersão da carga positiva
Porque é que os grupos alquilo estabilizam a carga positiva? Os grupos alquilo diminuem a concentração de carga positiva no carbocatião; A estabilidade aumenta com a capacidade de os substituintes “doarem” electrões ao carbono carregado Dispersão da carga positiva A estabilidade de um carbocatião depende dos grupos substituintes : os grupos alquilo cedem mais facilmente electrões por indução do que os H’s + A estabilização ocorre por polarização da nuvem electrónica da ligação s C-R: estabilização por indução

8
Porque é que os grupos alquilo estabilizam a carga positiva?
Uma carga positiva num carbono implica uma orbital p vazia, tal como no caso do catião metilo A presença de uma ligação s adjacente, por exemplo de uma ligação carbono-hidrogénio, possibilita a sobreposição de orbitais e consequente movimento dos electrões para a orbital p vazia A estabilização ocorre por deslocalização dos electrões por sobreposição de orbitais: estabilização por hiperconjugação

9
Adição electrofílica de HX
Postulado de Hammond: o estado de transição é estruturalmente semelhante às espécies que lhe estão mais próximas em termos de energia Adição electrofílica de HX Ep R2CH─CH2 + X- R2C─CH3 + X- RCH=CH2 + HX terciário primário Os factores que estabilizam os carbocatiões, também estabilizam os estados de transição A reacção ocorre no sentido em que se forma o carbocatião mais estável R2C─CH3 X R2CH─CH2 X

10
Adição electrofílica de HX
+ Uma reacção onde dois ou mais isómeros estruturais podem ser obtidos como produtos, mas forma-se predominantemente ou mesmo exclusivamente um deles é designada por reacção regiosselectiva. Podemos ter diferentes graus de regiosselectividade: moderada, alta ou completa No caso em que ocorre regiosselectividade completa só se forma um dos possíveis produtos, nos outros casos resultam misturas onde predomina um dos produtos

11
Adição electrofílica de HX
Para prever o produto de uma reacção de adição electrofílica a alcenos assimétricos, teremos de determinar as estabilidades relativas dos possíveis carbocatiões Em 1870 Markovnikov publicou um artigo descrevendo uma forma mais rápida de prever o produto da reacção Adição electrofílica de HX Vladimir V. Markovnikov ( ) Regra de Markovnikov: quando um alceno substituído de forma assimétrica reage com HX o átomo de hidrogénio é adicionado ao carbono que tiver maior número de hidrogénios, e o haleto ao que tiver menor número de hidrogénios Regra de Markovnikov: o electrófilo adiciona-se ao carbono sp2 que está ligado a mais hidrogénios
 Regra de Markovnikov: quando um alceno substituído de forma assimétrica reage com HX o átomo de hidrogénio é adicionado ao carbono que tiver maior número de hidrogénios, e o haleto ao que tiver menor número de hidrogénios. Regra de Markovnikov: o electrófilo adiciona-se ao carbono sp2 que está ligado a mais hidrogénios.")
12
C C C H X H OSO3H Adição electrofílica Segue a regra de Markovnikov,
Origina um hidrogenossulfato de alquilo

13
HOSO2OH CH3CH CH2 CH3CHCH3 OSO2OH Mecanismo + CH3CH CH2 H SO2OH O +
Adição electrofílica de H2SO4 HOSO2OH CH3CH CH2 CH3CHCH3 OSO2OH Hidrogenossulfato de isopropilo Mecanismo + CH3CH CH2 H SO2OH O lento + CH3CH CH3 SO2OH – O rápido CH3CHCH3 OSO2OH

14
H—OH CH3CHCH3 O SO2OH + D HO—SO2OH + CH3CHCH3 O H
Adição electrofílica de H2SO4 Os hidrogenossulfatos de alquilo são facilmente convertidos em álcoois por aquecimento em presença de água: hidrólise H—OH CH3CHCH3 O SO2OH + D HO—SO2OH + CH3CHCH3 O H

15
1. H2SO4 OH 2. H2O, D (75%) R2C=CH2, R2C=CRH, e R2C=CR2
Adição electrofílica de H2SO4 Os hidrogenossulfatos de alquilo são usados como intermediários na preparação de álcoois a partir de alcenos 1. H2SO4 2. H2O, D (75%) OH H2C=CH2, RCH=CH2, e RCH=CHR' Alcenos que originam hidrogenossulfatos de alquilo R2C=CH2, R2C=CRH, e R2C=CR2 Alcenos que não originam hidrogenossulfatos de alquilo
 OH. H2C=CH2, RCH=CH2, e RCH=CHR Alcenos que originam hidrogenossulfatos de alquilo. R2C=CH2, R2C=CRH, e R2C=CR2. Alcenos que não originam hidrogenossulfatos de alquilo.")
16
Adição electrofílica C H—X C X X—X C H X H—OSO3H H—OH C H OSO3H C H OH

17
C C X Adição de halogénios
X—X Esta reacção é limitada aos halogénios Cl2 e Br2 A utilização do F2 não é aconselhável dado ser explosivo A utilização do I2 não é eficiente, os di-iodetos vicinais sofrem decomposição libertando iodo e o alceno correspondente Di-halogeneto vicinal Efeito da estrutura do alceno Dado o seu efeito doador, quanto maior o número de substituintes maior a reatividade do alceno O X2 é apolar, mas facilmente polarizável; pode sofrer ataque nucleofílico pela dupla ligação do alceno Etileno H2C=CH2 1 Propeno CH3CH=CH2 61 2-metilpropeno (CH3)2C=CH 2,3-dimetil-2-buteno (CH3)2C=C(CH3)
2C=CH ,3-dimetil-2-buteno (CH3)2C=C(CH3)")
18
trans-1,2-dibromociclopentano
Adição de halogénios Br H Br2 H trans-1,2-dibromociclopentano A adição de X2 é estereosselectiva, ocorrendo em posição anti Br2 Br 1,2-dibromo-3-metilbutano Na adição de X2 não ocorrem rearranjos

19
Mecanismo Adição de halogénios
Um intermediário deste tipo não explica a adição anti, nem o facto de não ocorrerem rearranjos dibrometo vicinal Ião bromónio O ião bromónio justifica os resultados obtidos

20
– Br Br + Br Adição de halogénios
O ataque do Br– pelo lado oposto ao da ligação C-Br do ião bromónio é responsável pelo facto de esta adição ser anti

21
+ HX Qual é o resultado destas reacções quando existe água no meio?
Adição de halogénios Qual é o resultado destas reacções quando existe água no meio? + X2 + H2O X OH C + HX Em presença de água, a reacção de alcenos com X2 origina halidrinas

22
-OH Qual é o resultado destas reacções quando existe água no meio?
Adição de halogénios Qual é o resultado destas reacções quando existe água no meio? -OH Cl2 Ião clorónio O ataque do OH– pelo lado oposto ao da ligação C-Cl do ião clorónio é responsável pelo facto de esta adição ser anti

23
Qual é o resultado destas reacções quando existe água no meio?
Adição de halogénios Qual é o resultado destas reacções quando existe água no meio? Aplicação da regra de Markovnikov: A reacção pode ser vista como uma adição de HO-―Br+. O Br adiciona ao carbono que tiver mais hidrogénios ligados. Mais estável

24
Oximercuriação-redução
Mais estável Não ocorrem rearranjos

25
? Qual é o resultado destas reacções quando existe um álcool no meio?
Oximercuriação-redução Esta reacção é uma forma de adicionar água a alcenos Esta hidratação obedece à regra Markovnikov Qual é o resultado destas reacções quando existe um álcool no meio? ?

26
H + O H2B BH2 – BH3 Hidroboração-oxidação
Esta reacção também é uma forma de adicionar água a alcenos Esta hidratação é anti-Markovnikov A Hidroboração é normalmente feita usando como reagentes: O Borano-tetra-hidrofurano (H3B-THF) + O BH3 – •• H2B H BH2 Diborano B2H6
 + O. BH3. – •• H2B. H. BH2. Diborano B2H6.")
27
+ H—BR2 C H BR2 H2O2, HO– H OH C BR2 Hidroboração-oxidação
Adição de um organoborano à dupla ligação + H—BR2 C H BR2 Oxidação O produto formado é oxidado em meio alcalino com formação do álcool correspondente H2O2, HO– H OH C BR2

28
trans-2-metilciclopentanol (86%)
Hidroboração-oxidação Estereosselectividade A hidroboração é estereoselectiva: origina apenas adição syn: ou seja H e OH são adicionados pela mesma face da dupla ligação: 1. B2H6 2. H2O2, NaOH CH3 H HO trans-2-metilciclopentanol (86%) Não ocorrem rearranjos
 Não ocorrem rearranjos.")
29
Anti-Markovnikov Markovnikov Hidroboração-oxidação Adição de HBr
Mais estável Mais estável Adição de HBr Adição de BH3

30
Mecanismo Hidroboração-oxidação
Ligação da orbital p livre do borano à orbital p do alceno formação de um complexo p

31
Mecanismo Hidroboração-oxidação
O complexo p evolui no sentido de formação de um organoborano

32
Mecanismo Hidroboração-oxidação
O borano liga-se ao átomo de carbono menos substituído deste facto resulta a adição anti-Markovnikov O hidrogénio que se liga ao outro carbono é cedido pelo borano, resultando dai o facto de a adição ser syn

33
Mecanismo Hidroboração-oxidação Formação do ião hidroperóxido
Ataque nucleofílico do hidroperóxido ao boro

34
Mecanismo Hidroboração-oxidação
Migração do Carbono do Boro para o Oxigénio Hidrólise da ligação B―O

35
Hidroboração-oxidação
Mecanismo

36
CH3CH2CH CH2 HBr CH3CH2CHCH3 CH3CH2CH2CH2Br Br Adição radicalar de HX
Verificou-se que por vezes a adição de HBr a alcenos ocorriam por uma via anti-Markovnikov Verificou-se que a adiçãode HBr anti-Markovnikov ocorre sempre que no meio reaccional há peróxidos : R─O─O─R Na ausência de peróxidos a reacção segue a regra de Markovnikov CH3CH2CH2CH2Br CH2 CH3CH2CH HBr Com peróxidos Br CH3CH2CHCH3 Sem peróxidos * Não se observa com HCl ou HI

37
Adição radicalar de HX Mecanismo 1. Iniciação

38
Mecanismo Adição radicalar de HX 2. Propagação
Dois mecanismos distintos carbocatiões radicais permitem obter derivados distintos de forma regiosselectiva A adição anti-Markovnikov deve-se à maior estabilidade do radical terciário

39
Adição radicalar de HX Mecanismo 3. Finalização

40
Estabilidade de radicais
radical metilo radical terciário radical secundário radical primário Estabilidade crescente

41
Hidrogenação catalítica
+ H—H C H s p Exotérmica DH° = –136 kJ/mol catalisada por metais finamente divididos (Pt, Pd, Rh, Ni) Na ausência do catalisador a reacção é muito lenta A hidrogenação é uma reacção de redução
 Na ausência do catalisador a reacção é muito lenta. A hidrogenação é uma reacção de redução.")
42
A reacção ocorre em meio heterogéneo
Hidrogenação catalítica Moléculas de H2 na superfície do metal Aproximação do alceno Adição de H2 ao alceno A reacção ocorre em meio heterogéneo

43
Hidrogenação catalítica
Estereoquímica Na hidrogenação os átomos de hidrogénio são transferidos a partir da superfície do catalisador anti syn A adição dá-se preferencialmente “do mesmo lado” da dupla: adição Syn

44
Hidrogenação catalítica
Estereoquímica CO2CH3 (Cis: 100%) H2, Pt H CO2CH3
 H2, Pt. H. CO2CH3.")
45
Epoxidação Em presença de um peróxiácido os alcenos dão origem a compostos heterocíclicos designados por epóxidos Mecanismo

46
A epoxidação é estereosselectiva
Cis-2,3-dimetiloxirano Trans-2,3 óxidos de alceno” “epóxialcanos”

47
Epoxidação A partir de halidrinas

48
Formação de diois A reacção pode ser usada para confirmar a presença de alcenos: solução purpura → pp castanho Rendimentos mais elevados

49
Ozonólise O Ozono: O3 O ozono é uma molécula polar, um electrófilo forte e um oxidante muito forte Promove a quebra das cadeias carbonadas pela dupla ligação Aplicações em síntese: aldeidos e cetonas Aplicações analíticas: identificação dos substituintes dos carbonos da ligação dupla (devido à electronegatividade do oxigénio)
")
50
Ozonólise Mecanismo Reacção do alceno com ozono
ozónido Reacção do alceno com ozono compostos carbonílicos Hidrólise do ozónido com formação dos compostos carbonílicos The alkene and ozone undergo a concerted cycloaddition The molozonide is unstable because it has two O–O bonds Ozonide is stable Alternativamente a clivagem do ozónido Pode ser levada a cabo com sulfureto de dimetilo Os aldeídos formados oxidam facilmente a ácidos carboxílicos. O Zn é utilizado para prevenir essa oxidação

51
Redução Reacções!! O H OH (84%) NaBH4 etanol Redução Oxidação
Adição nucleófila O H OH (84%) NaBH4 etanol
 NaBH4. etanol.")
52
Oxidação RCR O [O] A oxidação de cetonas não ocorre nestas condições
![Oxidação RCR O [O] A oxidação de cetonas não ocorre nestas condições](http://slideplayer.com.br/slide/10386196/33/images/52/Oxida%C3%A7%C3%A3o+RCR+O+%5BO%5D+A+oxida%C3%A7%C3%A3o+de+cetonas+n%C3%A3o+ocorre+nestas+condi%C3%A7%C3%B5es.jpg "Oxidação RCR O [O] A oxidação de cetonas não ocorre nestas condições")
53
Oxidação Reagente de Tollens

54
Oxidação de Baeyer-Villiger
R"COOH O RCR' + R"COH O + ROCR' Ester

55
Oxidação O O C6H5COOH CCH3 OCCH3 CHCl3 (67%)
A inserção do O ocorre sempre entre o grupo mais substituído e o carbonilo Mecanismo O grupo que migra é o grupo mais substituído. Deste modo, a “aptidão migratória” segue a sequência seguinte: terciário > secundário > primário > metilo

56
Reactividade de aldeídos e cetonas
> Reactividade aumenta >

57
Reacções de adição nucleófila Mecanismo em meio básico

58
Reacções de adição nucleófila Mecanismo em meio ácido

59
São estas as reacções de adição nucleófila que vamos estudar
Adição de água Adição de reagentes de Grignard Adição de ácido cianídrico Adição de derivados de amoníaco Adição de álcoois

60
OH R R' C + H2O O C=O hidrato K % CH2=O CH2(OH)2 41 99.96 Hidratação
Efeito dos substituintes: Efeito Indutivo: os grupos alquilo estabilizam os reagentes grupos sacadores de electrões instabilizam o carbonilo Impedimento estereoquímico: os grupos alquilo instabilizam preferencialmente o derivado hidratado C=O hidrato K % CH2=O CH2(OH) CH3CH=O CH3CH(OH) (CH3)3CCH=O (CH3)3CCH(OH) (CH3)2C=O (CH3)2C(OH) Nas cetonas a extensão do equilíbrio de hidratação é muito menor do que nos aldeídos
 CH3CH=O CH3CH(OH) (CH3)3CCH=O (CH3)3CCH(OH) (CH3)2C=O (CH3)2C(OH) Nas cetonas a extensão do equilíbrio de hidratação é muito menor do que nos aldeídos.")
61
C O H – + HO C O – O H + O H – HO C OH •• • • • • •• +
E agora o mecanismo? + •• • • O H – HO C OH

62
C O + H + C OH H O + H O C OH H O O H C OH • • • • •• ••
E agora o mecanismo? • • O H •• C OH

63
d+ Et2O d– R’ R’ C C + O MgX MgX O – H3O+ R’ C OH
Reagentes de Grignard MgX + Et2O – O •• • • R’ C R1 R2 R’ MgX d– d+ • • O C •• R1 R2 H3O+ •• R’ C OH • • R1 R2

64
Reagentes de Grignard

65
Síntese de cianidrinas
+ •• • • C O HCN H C O N • • •• •• • • C O – N + – O N C •• • • H + O • • - H2O O N C •• • • H

66
Reacção com amoníaco e derivados Em certos casos temos catálise ácida
+ H2N R • • C O •• H C O HN R •• + H2O N R C •• Em certos casos temos catálise ácida A catálise ácida é importante para protonar o carbonilo Mas não deve protonar o derivado de amoníaco

67
C O H + C O + H+ C O N • • H + R + H2N R C O N • • H R •• •• • • • •
E agora o mecanismo? -H+ C O •• N • • H R

68
C O N H R C O N • • H R + H+ C N R H N R C •• •• + -H2O
E agora o mecanismo? N R C •• -H+

69
E agora o mecanismo?

70
Mecanismo em meio ácido
Protonação do oxigénio do grupo carbonilo, seguida do ataque da molécula do derivado de amoníaco Eliminação de água Eliminação de um protão

71
Mecanismo em meio neutro
Protonação o ataque ao carbono do grupo carbonilo Eliminação de água Eliminação de um protão

72
Reacção com amoníaco e derivados
+ H2N G • • C O •• + H2O N G C •• H R OH NH2 NHPh NHCONH2 G amoníaco (NH3) amina primária (NH2R) hidroxilamina (NH2OH) hidrazina (NH2NH2) fenil-hidrazina (NH2NHPh) semicarbazida (NH2NHCONH2) Reagente a utilizar
 amina primária (NH2R) hidroxilamina (NH2OH) hidrazina (NH2NH2) fenil-hidrazina (NH2NHPh) semicarbazida (NH2NHCONH2) Reagente a utilizar.")
73
Qual será a estrutura dos produtos? E o nome da família?
hidrazona iminas oxima semicarbazola fenil-hidrazona

74
Adição de álcoois

75
C R R' O 2R"OH + OR" + H2O Adição de álcoois
A reacção de formação de acetais é reversível O sentido em que a reacção se dá depende do controle adequado da quantidade de álcool e de água no sistema Podem ser usados como grupos protectores dos carbonos carbonílicos em reacções em que na forma livre estes podiam ser transformados

76
C O H + C O + H+ R H O C O • • H + R C O • • H R •• •• • • + • •
E agora o mecanismo? C O •• • • H R -H+ Hemi-acetal

77
C O • • H R + H+ C O H R + C O R + C O R + •• •• E agora o mecanismo?
-H2O E agora o mecanismo? C O R •• + C O •• R + Carbocatião estabilizado por ressonância

78
C O R + C O R + H R H O C O R Acetal •• •• + -H+ • • ••
E agora o mecanismo? Acetal

79
Agora vamos resolver alguns exercícios

80
Agora vamos resolver alguns exercícios

81
Agora vamos resolver alguns exercícios

82
Equilíbrio ceto-enólico
Em geral a forma cetónica é muito mais estável que a forma enólica Excepções: O enol é estabilizado por uma ponte de hidrogénio intramolecular

83
meio alcalino meio ácido
E o mecanismo? meio ácido

84
Acidez do protão a carbono a hidrogénio a
anião estabilizado por ressonância

85
Baixas temperaturas (0-5 oC) => reacção termina no aldol
CONDENSAÇÃO ALDÓLICA Baixas temperaturas (0-5 oC) => reacção termina no aldol Temperaturas mais elevadas => resulta o composto a,b-insaturado A conjugação entre a ligação dupla e o carbonilo torna o composto mais estável A condensação aldólica pode também ser conduzida em meio ácido
 => reacção termina no aldol. Temperaturas mais elevadas => resulta o composto a,b-insaturado. A conjugação entre a ligação dupla e o carbonilo torna o composto mais estável. A condensação aldólica pode também ser conduzida em meio ácido.")
86
H O Formação do enolato Ataque nucleofílico a C=O E agora o mecanismo?
-OH- O H • • •• aldol

87
Desidratação -H2O E agora o mecanismo?

89
Condensação aldólica cruzada
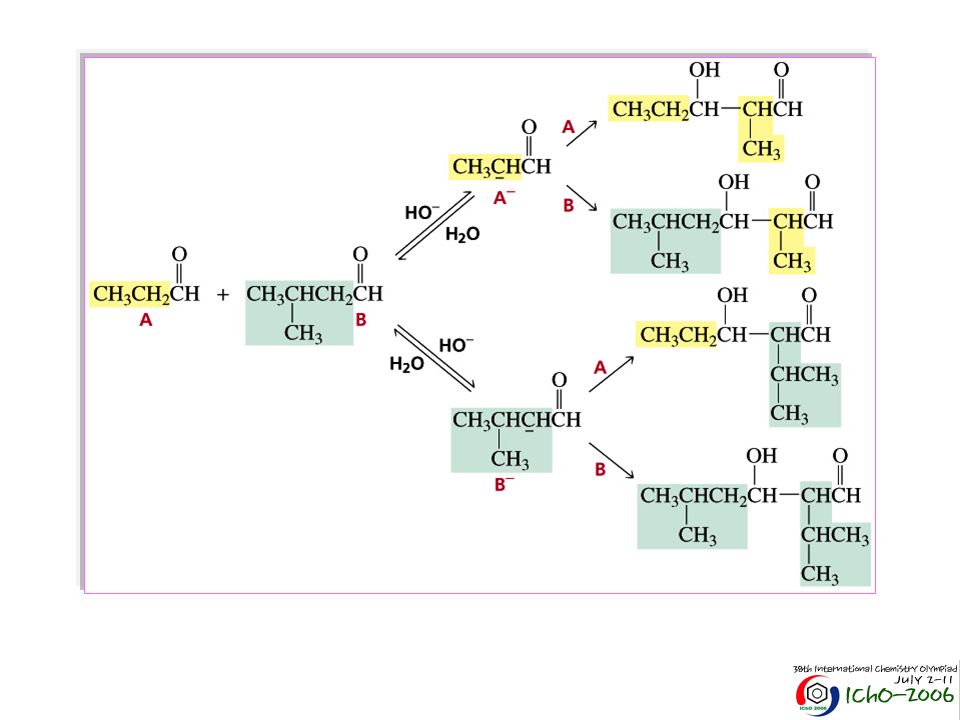
Há que minimizar o número de possibilidades Usar apenas um componente que forme enolato CH3O CH O + CH3CCH3 CH3O CH CHCCH3 O (83%) NaOH, H2O 30°C
 NaOH, H2O. 30°C.")
90
Reacção de Cannizzaro Aldeídos sem hidrogénios em posição a podem sofrer auto- -oxidação e auto-redução, quando estão em presença de soluções alcalinas concentradas

91
Mais um mecanismo!

92
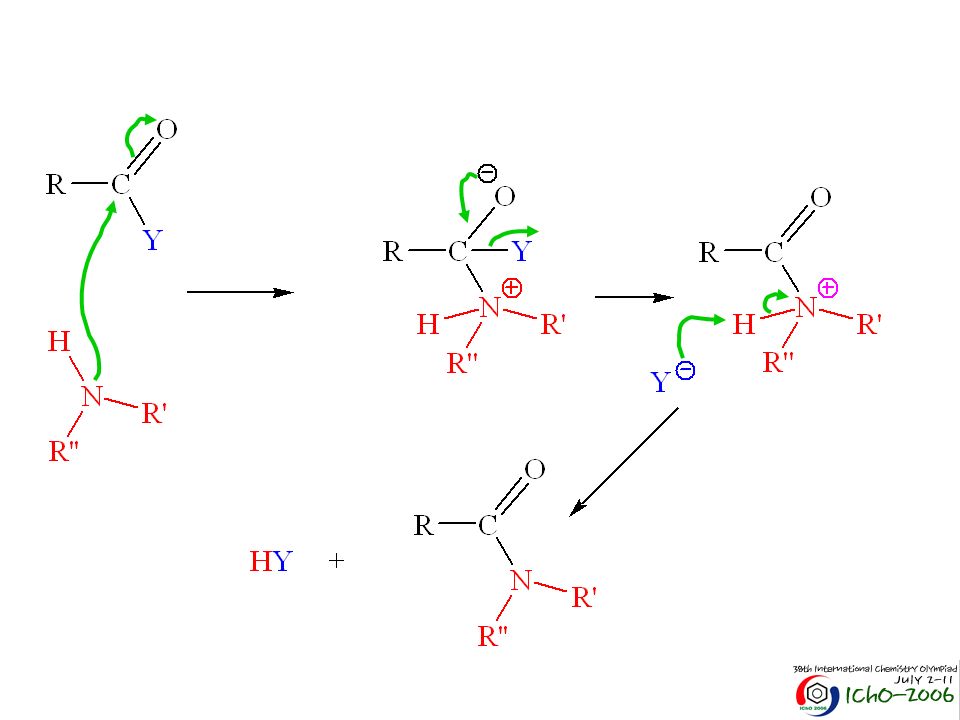
intermediário tetraédrico adição nucleofílica eliminação Nos ácidos carboxílicos temos reacções de substituição nucleofílica. Como a reacção ocorre no carbono “acílico” é designada por substituição acílica. base mais fraca despede-se mais facilmente

93
B- + HNu Nu- + HB Y- + HB B- + HY
Este mecanismo descreve uma catálise básica: podemos ter uma base para facilitar a formação da espécie nucleofílica no final a espécie que se despede pode captar o protão e libertar a base B HNu Nu HB Y HB B HY

94
Porquê? Nos ácidos temos:
Ora nos aldeídos e cetonas tinhamos uma reacção de adição nucleofílica. Agora nos ácidos carboxílicos temos uma substituição nucleofílica. Porquê? Nos ácidos temos: Geometria trigonal planar Geometria trigonal planar Intermediário tetraédrico instável Maior proximidade dos grupos Pode eliminar OH-

95
Nos aldeídos e cetonas temos:
Ora nos aldeídos e cetonas tinhamos uma reacção de adição nucleofílica. Agora nos ácidos carboxílicos temos uma substituição nucleofílica. Porquê? Nos aldeídos e cetonas temos: Geometria trigonal planar Geometria tetraédrica Intermediário tetraédrico instável Maior proximidade dos grupos Não pode eliminar H- ou R-

96
A substituição nucleofílica pode ocorrer por catálise ácida

97
Reactividade relativa dos derivados de ácidos carboxílicos

98
RCO– O Cl – OR NH2 < RC O X + – • • ••
A basicidade do grupo que é eliminado RCO– O Cl – OR NH2 < Estabilidade do derivado RC O X •• • • + –

99
Reacção com reagentes de Grignard
Os ésteres e os cloretos de acilo por reacção com duas moles de reagente de Grignard seguida de um tratamento com ácido diluído produzem álcoois

100
– MgX + R C O OCH3 R' C O d+ OCH3 R' R MgX d– Et2O –CH3OMgX C O R R'
Mecanismo – MgX + R C O •• • • OCH3 R' C O •• • • d+ OCH3 R' R MgX d– Et2O –CH3OMgX C O R R' • • •• O intermediário formado é instável e elimina o grupo alcóxido formando a cetona correspondente

101
Descarboxilação de ácidos carboxílicos
Reacção que ocorre com muita dificuldade No entanto, quando temos um b-cetoácido a descarboxilação ocorre com facilidade

102
Descarboxilação de ácidos carboxílicos
Descarboxilação do anião carboxilato

103
Descarboxilação de ácidos carboxílicos
Descarboxilação do ácido

104
Reacção de Hell-Volhard-Zelinskiy
Ácido a-haloalcanóico Haleto de a-haloacilo

105
R2C H C O OH R2C H C O X PX3 R2C H C O X Mecanismo
Formação do haleto de acilo R2C H C O OH R2C H C O X PX3 Formação da forma enólica do haleto de acilo R2C H C O X

106
R2C H C O X R2C Y C O X Y R2C- Y C- O OH - HY H2O Mecanismo
Halogenação da forma enólica R2C H C O X R2C Y C O X - HY Y Regeneração do grupo carboxilo H2O R2C- Y C- O OH

107
Na síntese de cloretos de acilo utilizam-se outros cloretos de acilo muito mais reactivos:
Cloreto de tionilo (SOCl2) Tricloreto de fósforo (PCl3) Pentacloreto de fósforo (PCl5)
 Tricloreto de fósforo (PCl3) Pentacloreto de fósforo (PCl5)")
108
Mecanismo

109
A hidrólise de cloretos de acilo
Mecanismo

110
Na síntese de anidridos de ácidos carboxílicos utilizam-se dois métodos:
Reacção de sais de sódio de ácidos carboxílicos com cloretos de acilo Reacção de ácidos carboxílicos com cloretos de acilo

111
Hidrólise de anidridos:

112
Mecanismo? Preparação de ésteres:
A partir de ácidos carboxílicos, usualmente designada por esterificação Catalisador: H2SO4 ou HCl A reacção directa é favorecida pela eliminação de água Mecanismo?

113
C O H + R H C O R + H C O H + R O CH3 H C OH O + CH3 H R + -H C OH O
•• C O H • • + R H C O •• • • R + H C O H • • + R • • O CH3 H C OH •• • • O + CH3 H R + -H •• C OH • • O CH3 R

114
C OH O R C OH O OCH3 H + R CH3 -H2O C OH OCH3 + R + H C O―H OCH3 + R R
•• C OH • • O R •• C OH O • • OCH3 H + R CH3 -H2O C OH •• • • OCH3 + R + H C O―H •• OCH3 + R R C O OCH3 •• • • + H

115
A partir de cloretos de acilo

116
A partir de anidridos de ácidos carboxílicos

117
A partir de outros ésteres, usualmente designada por transesterificação
Removendo, por destilação, o álcool R’OH desloca-se o equilíbrio no sentido da transesterificação. No entanto, é necessário que este álcool tenha um ponto de ebulição menor do que o do álcool R’’OH Qual será o mecanismo?

118
Desta vez não nos livramos do mecanismo!
A partir de cetonas, por oxidação de Baeyer-Villiger Desta vez não nos livramos do mecanismo!

119
Lactonas: ésteres especiais
Os ácidos carboxílicos g e d hidroxilados podem ter uma reacção de esterificação intramolecular, a qual ocorre em meio ácido. Desta reacção resulta a formação de g- ou d-lactonas

120
Hidrólise de ésteres: Hidrólise alcalina (saponificação) Hidrólise ácida
 Hidrólise ácida")
121
E agora os mecanismos!

122
E agora os mecanismos!

123
E agora os mecanismos!

124
Preparação de amidas - Amonólise
E agora o mecanismo!

126
A partir de ácidos carboxílicos:
Preparar o sal correspondente Secar e aquecer a altas temperaturas => ocorre desidratação e consequente formação da amida Não é um método muito eficiente

127
Hidrólise de amidas: Hidrólise alcalina Hidrólise ácida

128
E agora os mecanismos!

129
E agora os mecanismos!

130
Lactamas g-lactama Formam-se b, g, d e e-lactamas, respectivamente com anéis de quatro, cinco, seis e sete lados As b-lactamas são muito reactivas

131
Resumindo De um modo geral é possível transformar um derivado noutro menos reactivo. A transformação inversa é muito difícil ou mesmo impossível. Reactividade relativa: cloreto de acilo > anidrido > éster > amida > ácido

132
Resumindo De um modo geral é possível transformar um derivado noutro menos reactivo. A transformação inversa é muito difícil ou mesmo impossível. Reactividade relativa: cloreto de acilo > anidrido > éster > amida > ácido

133
Resumindo De um modo geral é possível transformar um derivado noutro menos reactivo. A transformação inversa é muito difícil ou mesmo impossível. Reactividade relativa: cloreto de acilo > anidrido > éster > amida > ácido

134
Resumindo De um modo geral é possível transformar um derivado noutro menos reactivo. A transformação inversa é muito difícil ou mesmo impossível. Reactividade relativa: cloreto de acilo > anidrido > éster > amida > ácido

Apresentações semelhantes




 C A +>")














