Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Disciplina de Genética e Evolução
SÍNDROME DE DIGEORGE Camila Furtado de Souza Erica Marquardt Lämmerhirt Cláudio Borba Canabarro Juliana Pereira Passaglia Monitor: André Luís Ferreira da Silva

2
GENERALIDADES A Síndrome de DiGeorge (DGS) caracteriza-se por uma série de mal-formações congênitas. defeitos cardíacos e dos grandes vasos; hipoplasia ou aplasia de timo e paratireóides; dismorfismos faciais.

3
A deleção de um gene ou de um grupo de genes adjacentes no cromossomo 22q11 (del22q11) é provavelmente a causa desta síndrome. Alterações em outras seqüências específicas do DNA também foram descritas. Agentes teratogênicos como potenciais causadores.

4
Síndromes de microdeleções ou Síndromes dos genes contíguos.
Síndromes de Deleção 22q11: Síndrome Velocardiofacial (VCFS) ou de Shprintzen; Síndrome Cardiofacial de Caylor; Síndrome de Opitz G/BBB; Síndrome CATCH-22.
 ou de Shprintzen; Síndrome Cardiofacial de Caylor; Síndrome de Opitz G/BBB; Síndrome CATCH-22.")
5
EPIDEMIOLOGIA Método de FISH n° de casos diagnosticados (20% através de análise citogenética). A deleção foi encontrada em 5% das crianças com defeitos cardíacos congênitos. Estimativa de incidência de pelo menos 1:4000 nascidos vivos. Mutação nova x padrão de herança familiar.

6
Dificuldades no aconselhamento genético.
- história familiar - defeitos cardíacos e/ou fissuras labiopalatinas à ecografia DGS relaciona-se à elevada morbimortalidade neonatal (defeitos cardíacos, imudeficiência e hipocalcemia). Manejo da criança sindrômica requer equipe multidisciplinar.
. Manejo da criança sindrômica requer equipe multidisciplinar.")
7
Fundamentos Genéticos
Deleção intersticial no braço longo do cromossomo 22 - del22q11 90% dos pacientes possuem deleções sobreponíveis em região de 2-3 Mb - Região Tipicamente Deletada (TDR)
")
8
Fundamentos Genéticos
Causas de deleção: pareamento desalinhado dos cromossomos homólogos na meiose I LCRs (low copy repeats) rearranjos intra e intercromossômicos segregação anormal a partir de uma translocação ou inversão
 rearranjos intra e intercromossômicos. segregação anormal a partir de uma translocação ou inversão.")
9
Fundamentos genéticos
Translocações balanceadas: ruptura de seqüência gênica DGS pais com translocação e não afetados segregação desequilibrada na gametogênese filhos com DGS

10
Padrão de Herança MUTAÇÃO NOVA Condição esporádica
pais não portadores, filhos afetados Risco de recorrência igual ao de população em geral (exceto no caso de mosaicismo germinativo)
")
11
Padrão de Herança Autossômico Dominante:
Um dos pais afetado % de chance de recorrência

12
Síndrome de Genes Contíguos ou Monogênica ???

13
Síndrome de genes contíguos
Vários genes envolvidos (25-40 genes na DGS) Fenótipo variável e complexo
 Fenótipo variável e complexo.")
14
Síndrome de genes contíguos
Argumentação: pacientes com fenótipo compatível com DGS apresentam deleções não sobreponíveis nenhum segmento da região TDR está deletado em todos os pacientes

15
Monogênica Proposta recente: 1 gene envolvido, com penetrância e expressividade variável gêmeos MZ com apresentação clínica discordante; tamanho da deleção não tem relação com a gravidade da apresentção clínica. PORÉM: não se identificou segmento deletado comum a todos os pacientes

16
EFEITO DE HAPLOINSUFICIÊNCIA
Patogênese hemizigose para Del22q genes da região deletada EFEITO DE HAPLOINSUFICIÊNCIA

17
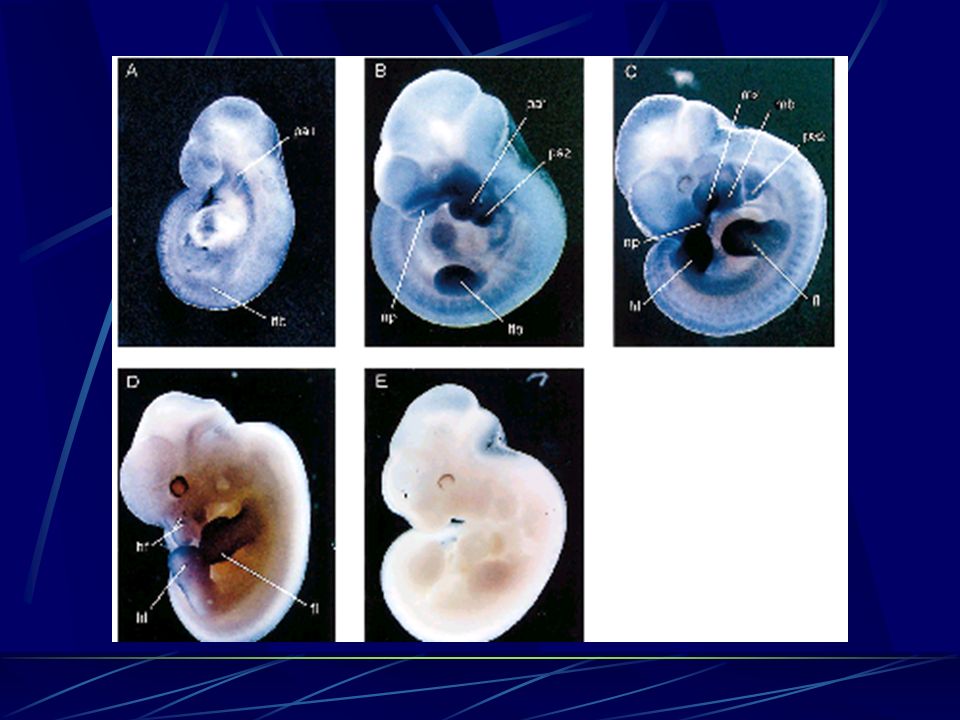
Déficit de migração das cel. da crista neural para bolsas faríngeas
Patogênese Distúrbio desenvolvimental - Embriogênese Haploinsuficiência Alteração da morfogênese dos órgãos: timo, paratireóides, via de saída cardíaca, estruturas craniofaciais Déficit de migração das cel. da crista neural para bolsas faríngeas

19
Quais genes envolvidos??
Os genes candidatos devem: 1) ser expressos na embriogênese, especialmente nos órgãos afetados na DGS; 2) participar do desenvolvimento das cel. da crista neural; 3) estar deletados nos indivíduos com DGS; 4) 1 ou mais pacientes com DGS apresentando deleção menor ou mutação puntiforme no gene candidato.
 ser expressos na embriogênese, especialmente nos órgãos afetados na DGS; 2) participar do desenvolvimento das cel. da crista neural; 3) estar deletados nos indivíduos com DGS; 4) 1 ou mais pacientes com DGS apresentando deleção menor ou mutação puntiforme no gene candidato.")
20
Genes candidatos COMT: enzima de degradação das catecolaminas
Alelo de baixa atividade Gene deletado degradação das catecolaminas Psicose (?)
")
21
Gene candidato ZNF74: codifica proteína com motivo em “zinc finger”
propriedade de se ligar ao DNA possível fator de transcrição; RNAm do ZNF74 presente em tecidos embrionários, mas não em tecidos adultos.

22
ZNF74 - proteínas com “zinc finger”

23
Gene candidato: TUPLE - 1
é um regulador transcricional em leveduras; função em humanos (?) nem todos os pacientes possuem deleção deste gene.
 nem todos os pacientes possuem deleção deste gene.")
24
Gene candidato - UFD1L + de 85% dos pacientes apresentam deleção;
codifica proteína ubiquitina de fusão-degradação; papel importante na formação do fenótipo de DGS;

25
Gene candidato - UFD1L Em leveduras:
- crescimento e diferenciação celular - transdução de sinal - tráfego transmembrana ativa na via de degradação proteolítica formação da cauda poli(A) do RNAm
 do RNAm.")
26
Gene candidato - UFD1L Em mamíferos:
possível papel no transporte nuclear; RNAm difundido e expresso na vida fetal e pós-natal.

27
Gene candidato - UFD1L Teorias:
haploinsuficiência do UFD1L levaria a acúmulo de proteínasAPOPTOSE PRECOCE das cel da crista neural; cel com uma única cópia do UFD1L seriam mais suscetíveis a outros insultos genéticos, ex: mutação em outro gene crítico da TDR.

28
Gene candidato - PCQAP Codifica subunidade proteica de complexo Coativador da transcrição dos genes de classe II; Papel na regulação da transcrição de genes de classe II;

29
Gene candidato - PCQAP Transcrição dos genes de classe II
Componentes: RNA pol II; GTFs (fatores de transcrição basais); Ativadores; Coativadores Controlam a resposta da maquinaria basal aos ativadores Facilitam a formação dos complexos de iniciação da transcrição
; Ativadores; Coativadores. Controlam a resposta da maquinaria basal aos ativadores. Facilitam a formação dos complexos de iniciação da transcrição.")
30
Coativadores da transcrição de genes de classe II

31
Gene candidato - PCQAP expressão na embriogênese
expressão na massa fronto nasal, bolsas faríngeas e brotos dos membros.

32
Gene candidato - Tbx1 É um fator transcricional da família T-box;
camundongos com deleção deste gene apresentaram: Gene em estudos hipoplasia de timo e de paratireóides; - alterações da via de saída cardíaca; - anomalias craniofaciais.

33
Questões Nenhum dos genes citados se encontra deletado em todos os pacientes: contribuição de muitos genes na formação da doença? mecanismos regulatórios de longa extensão na TDR? como explicar variabilidade genótipo-fenótipo?

34
ASPECTOS CLÍNICOS Principais anormalidades: - tímicas - paratireóideas
- cardíacas - faciais

35
A) TÍMICAS: Imunidade celular afetada Síndrome parcial Síndrome completa
 TÍMICAS: Imunidade celular afetada Síndrome parcial Síndrome completa")
36
B) PARATIREÓIDEAS: Deficiência de PTH Tetania / convulsões
 PARATIREÓIDEAS: Deficiência de PTH Tetania / convulsões")
37
C) CARDÍACAS: Tetralogia de Fallot Tronco arterioso Arco aórtico com implantação à direita Mal-formações valvares, arteriais, miocárdicas Defeitos de septo interventricular / interatrial

38
D) FACIAIS: Hipertelorismo Orelhas com baixa implantação Fissura palatina Micrognatia Úvula bífida
 FACIAIS: Hipertelorismo Orelhas com baixa implantação Fissura palatina Micrognatia Úvula bífida")
39
ANOMALIAS FACIAIS

40
OUTRAS: Mal-formações das vias aéreas Distúrbios auditivos / oculares Anormalidades esofágicas / tireoidianas Distúrbios psiquiátricos Déficit de crescimento Dificuldade no aprendizado Defeitos no trato urinário

41
Diagnóstico Diagnóstico Clínico:
Manifestações decorrentes da aplasia tímica; Hipoparatireoidismo; Mal-formações cardíacas Fácies dismórfica Déficit neurológico.

42
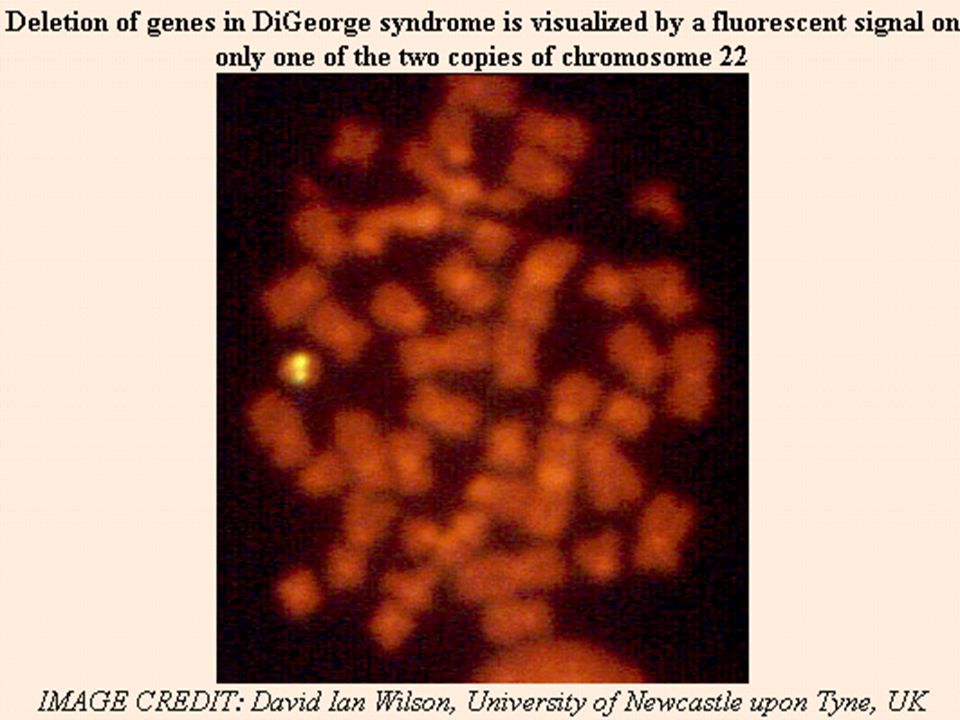
Diagnóstico Diagnóstico Citogenético:
Técnicas de bandeamento cromossômico; Técnicas citogenéticas moleculares: FISH PCR

43
Diagnóstico - FISH

45
Diagnóstico Diagnóstico Pré-Natal: Aplicabilidade Métodos
Aconselhamento pré-natal

46
Diagnóstico Diagnóstico Diferencial: Anomalias cardíacas congênitas
Hipocalcemia inexplicada Infecções de repetição

47
MANEJO DA CRIANÇA PORTADORA DE DGS
Necessidade de equipe multidisciplinar. Tratamento individualizado. Inicialmente: - restauração da competência imune e dos níveis séricos de cálcio. - planejamento seqüencial.

48
Período neonatal: dosagem sérica de cálcio - concentrações requerem reposição - tetania nas 1as h de vida contagem absoluta dos linfócitos - linfopenia: avaliação dos subtipos de céls. T e B - evitar vacinas vivas atenuadas

49
Imunodeficiência: > susceptibilidade a infecções história de infecções severas de repetição transplante de tecido tímico pode restaurar a função imune Ainda no período neonatal: - ultrassom renal - avaliação cardiológica

50
Avaliação endocrinológica:
Hipoparatireoidismo Déficit de crescimento ( GH?) Hipertireoidismo Trato Gastrointestinal: Dificuldade de alimentação Refluxo gastroesofágico Acompanhamento fonoaudiológico.
 Hipertireoidismo. Trato Gastrointestinal: Dificuldade de alimentação. Refluxo gastroesofágico. Acompanhamento fonoaudiológico.")
51
Prognóstico variável:
- pacientes que recuperam espontaneamente a atividade do sistema imune; - pacientes praticamente assintomáticos. Suporte psicológico.

Apresentações semelhantes