Carregar apresentação
A apresentação está carregando. Por favor, espere

1
PROPEDÊUTICA ESTOMATOLÓGICA
Leandro Flores Nogueira Prof. Ms. em Ciências Médicas ICT – CURSO DE ODONTOLOGIA Novembro/2015

2
Neoplasias Hematológicas:
Leucemias Linfomas Gamapatias Monoclonal Histiocitose X (Doença das Células de Langerhans)
")
5
Leucemias Definição Proliferação neoplásica generalizada de células
hematopoiéticas oriundas de um mesmo clone (monoclonal). Doenças clonais, originadas por uma célula tronco hematopoiética pluripotente, a qual sofreu uma alteração genética.
. Doenças clonais, originadas por uma célula tronco. hematopoiética pluripotente, a qual sofreu uma. alteração genética.")
6
Etiologia Leucemias Não está totalmente esclarecida
Acredita-se que alterações citogenéticas sejam resultantes de um ou mais determinantes ambientais que atuem em um indivíduo particularmente susceptível

7
Fatores desencadeantes:
Leucemias Fatores desencadeantes: AMBIENTE: radiação ionizante (agente leucemogênico) drogas e agentes químicos: (cloranfenicol, fenilbutazona, benzeno) vírus (HTLV I e HTLV II) HOSPEDEIRO fatores genéticos (vários casos na família, imunodeficiência e disfunção da MO)
 drogas e agentes químicos: (cloranfenicol, fenilbutazona, benzeno) vírus (HTLV I e HTLV II) HOSPEDEIRO. fatores genéticos. (vários casos na família, imunodeficiência e disfunção da MO)")
8
Leucemias Classificação Leucemias agudas: infiltrado medular com predominância de blastos, curso clínico agressivo Leucemias crônicas: maior proporção de células diferenciadas ou madura, clinicamente tem início insidioso

9
Leucemias Leucemias Mielóides Aguda – M0 até M7 Crônica Leucemias Linfóides - Aguda – L1 até L3 - Crônica

11
Leucemias Leucemias agudas: - curso rápido, pode ser fatal células blásticas da série mielóide ou linfóide (MO ou sangue periférico) mais de 20% de blastos na MO - (OMS) Manifestações clínicas: anemia, (palidez e fraqueza) infecção e febre (granulocitopenia) púrpuras e hemorragias (plaquetopenia) esplenomegalia, dores ósseas e reumáticas (infiltração de células em diversos órgãos)
 Manifestações clínicas: anemia, (palidez e fraqueza) infecção e febre (granulocitopenia) púrpuras e hemorragias (plaquetopenia) esplenomegalia, dores ósseas e reumáticas. (infiltração de células em diversos órgãos)")
12
Diagnóstico: anemia (normocítica e normocrômica) presença de eritroblastos trompocitopenia (inicialmente ou tardiamente) leucocitose com presença de blastos adulto (LMA) crianças (LLA)
 crianças (LLA)")
13
O primeiro sinal ou sintoma de leucemia ocorre na boca (Weckx et al
O primeiro sinal ou sintoma de leucemia ocorre na boca (Weckx et al.1990), sendo mais comum nas fases agudas da doença (Shafer et al., 1987), e mais frequentes na leucemia monocítica (Burkitt)
, sendo mais comum nas fases agudas da doença (Shafer et al., 1987), e mais frequentes na leucemia monocítica (Burkitt)")
14
As lesões primárias que ocorrem na gengiva são caracterizadas por aumento da papila interdental e da gengiva marginal. A gengiva fica friável, sangra com facilidade e podem ocorrer infecções secundárias. Podem ocorrer gengivite, hiperplasia gengival, hemorragia, petéquias e ulceração de mucosa no palato, assoalho oral e língua.

16
Tratamento Visa eliminar o clone maligno e repopular a MO com células normais Estratégias de tratamento: quimioterapia, radioterapia e transplante de MO Terapia suporte (transfusão): concentrado glóbulos vermelhos (anemia) - concentrado plaquetário (trombocitopenia)
: concentrado glóbulos vermelhos (anemia) - concentrado plaquetário (trombocitopenia)")
17
Leucemia Linfóide Aguda - (LLA)
acomete principalmente crianças (adultos também) LLA pode ser do tipo B (mais freqüente) ou tipo T presença de linfoblastos classificação FAB (L1 até L3) prognóstico: crianças: 2-10 anos (melhor recuperação)
 LLA pode ser do tipo B (mais freqüente) ou tipo T. presença de linfoblastos. classificação FAB (L1 até L3) prognóstico: crianças: 2-10 anos (melhor recuperação)")
18
Mal prognóstico: sexo masc. raça negra, leucocitose, infiltração do SNC e tipo celular L3 tratamento: drogas: quimioterápicas: Prednisona, Vincristina, L-asparaginase e Daunomicina cura: 4 a 5 anos em remissão completa

19
Leucemia Mielóide Aguda - (LMA)
proliferação anormal dos precursores mielóides com perda da diferenciação e maturação celular classificação de FAB – M0 até M7 (baseada na natureza dos blastos e no grau de diferenciação e maturação)
")
20
- mais freqüente – sexo masculino
- bastões de AUER em alguns blastos (prova peroxidase e sudan back – positivas) - tratamento drogas quimioterápicas associadas a (Citarabina (ARA-C), Daunomicina e Tioguanine) transplante MO (30 a 50% cura), quando encontrado doador compatível
 - tratamento drogas quimioterápicas associadas a. (Citarabina (ARA-C), Daunomicina e Tioguanine) transplante MO (30 a 50% cura), quando encontrado doador. compatível.")
21
Leucemias crônicas proliferação células madura (linfóide ou mielóide) atinge idade média ou senil (sexo masculino mais comum) inicialmente assintomática, anorexia, fraqueza, emagrecimento, anemia achados clínicos: esplenomegalia (leucemia mielóide), adenopatia (leucemia linfóide) e * anemia em ambos os tipos
, adenopatia (leucemia linfóide) e. * anemia em ambos os tipos.")
22
Leucemias Leucemia Linfóide Crônica - (LLC) proliferação de células linfóides maduras disfuncionais doença lenta e progressiva (rara antes do 40 anos) - anomalias cromossômicas mais frequentes trissomia 12 (+12q) e a deleção do 13 (-13q14) Classificadas em: tipo B (CD19, CD20 positivos) maioria - tipo T (CD2, CD3, CD4 e CD5 positivos ) 10 % apenas
 - anomalias cromossômicas mais frequentes. trissomia 12 (+12q) e a deleção do 13 (-13q14) Classificadas em: tipo B (CD19, CD20 positivos) maioria. - tipo T (CD2, CD3, CD4 e CD5 positivos ) 10 % apenas.")
23
Diagnóstico tumores linfóides – adenopatias indolores, esplenomegalia, Hemograma: leucocitose variável a / mm3 (linfócitos maduros, prolinfócitos, raramente linfoblastos) anemia, trombocitopenia, granulocitopenia mielograma
 anemia, trombocitopenia, granulocitopenia. mielograma.")
24
Tratamento: varia de acordo com o grau de estadiamento da doença quimioterapia nos casos de tumores linfóides e insuficiência medular grave Clorambucil, Ciclofosfamida, Prednisona - transplante MO – para pacientes mais jovens

25
Leucemia Mielóide Crônica (LMC)
- 1ª neoplasia descrita por ativação de proto-oncognes celulares 95% dos pacientes LMC, apresentam cromossomo Philadelfia (cromossomo 22 anormal oriundo da translocação (9;22) (q34; q11) proto-oncogene c-abl (cromossomo 9 é transferido para o cromossomo 22 ligando-se ao gene bcr) O gene híbrido (bcr/abl) -> codifica proteína com atividade tirosina quinase aumentada -> interfere no ciclo celular -> proliferação e diferenciação descontrolada das células leucêmicas
 (q34; q11) proto-oncogene c-abl (cromossomo 9 é transferido para o. cromossomo 22 ligando-se ao gene bcr) O gene híbrido (bcr/abl) -> codifica proteína com atividade tirosina quinase aumentada. -> interfere no ciclo celular. -> proliferação e diferenciação descontrolada das células leucêmicas.")
26
- Crise blástica: (transformação aguda) – 30% de bastos MO
LMC - 3 fases: - Crônica: duração de 3 anos hiperleucocitose, células mielóides maduras - Acelerada: células perdem gradualmente a capacidade de diferenciação. Presença de basofilia, trombocitose, anemia. - Crise blástica: (transformação aguda) – 30% de bastos MO ou sangue periférico (sobrevida de 2 a 10 meses) * rara em crianças / comum em adultos jovens e meia idade
 – 30% de bastos MO. ou sangue periférico (sobrevida de 2 a 10 meses) * rara em crianças / comum em adultos jovens e meia idade.")
27
Quadro clínico Instalação insidiosa, descoberta acidental, mal estar, perda de peso ou febre esplenomegalia, associada ou não a hepatomegalia Diagnóstico hemograma: hiperleucocitose a acima de /mm3 (predomínio série granulocítica) - pseudo desvio a esquerda (sem ordem de maturação das células) - frequente basofilia e e eosinofilia - anemia (maioria dos casos) - trombocitose ( 50%) - trombocitopenia (15%)
 - pseudo desvio a esquerda (sem ordem de maturação das células) - frequente basofilia e e eosinofilia. - anemia (maioria dos casos) - trombocitose ( 50%) - trombocitopenia (15%)")
28
Diagnóstico mielograma: hiperplasia granulocítica (80 a 95% )
citogenética: presença do cromossomo Philadelfia (mais de 95% dos indivíduos) - biologia molecular (ou análise molocular): teste diferencial para detecção do gene bcr-abl (gene híbrido).
 - biologia molecular (ou análise molocular): teste diferencial para detecção do gene bcr-abl (gene híbrido).")
30
Tratamento - mielossupressão com quimioterápicos: Bussulfan e drogas inibidoras da síntese de DNA (Hidroxiuréia) - Interferon α - supressão do cromossomo Philadelfia Gleevec: inibi a proliferação do clone leucêmico, induzindo a apoptose (não interfere nas células normais) - transplante MO (única possibilidade de cura; idade do paciente, doador compatível, indicada na fase crônica - 1º ano de diagnóstico)
 - transplante MO. (única possibilidade de cura; idade do paciente, doador. compatível, indicada na fase crônica - 1º ano de diagnóstico)")
31
Síndrome Mielodisplásica

32
Linfomas

33
Conceito Neoplasias malignas derivadas da expansão de um clone de células linfóides O estágio de maturação celular em que ocorre a transformação maligna determina o surgimento de diferentes tipos de linfomas, com características morfológicas, moleculares e biológicas distintas.

34
Epidemiologia Os linfomas representam o terceiro grupo mais comum de lesões malignas da cavidade bucal e região maxilofacial, seguindo as lesões de carcinoma de células escamosas e de glândulas salivares. Na boca, os linfomas são LNH e acometem mais frequentemente a gengiva, palato e língua. Clinicamente, os linfomas de boca apresentam-se como massas de tecido mole, firmes e podem ser indolores ou apresentarem sintomatologia. Os linfomas de boca podem tornar-se ulcerados e assim semelhantes ao carcinoma de células escamosa de boca.

35
Classificações dos Linfomas
Rappaport Lukes e Collins Kiel REAL OMS

36
Classificação dos Linfomas - OMS
Linfomas não Hodgkin Neoplasias de células linfóides B Precursoras Maduras Neoplasia de células T/NK Linfoma de Hodgkin

37
Linfomas não Hodgkin Grupo heterogêneo de neoplasias que acomete pacientes de todas as idades. 30 subtipos histológicos, em sua maioria de origem B Altamente sensíveis a QT Acometem os gânglios linfáticos, baço, fígado e medula óssea, mas em 25% dos casos tem apresentação extra-nodal: estômago, pele, cavidade oral, intestino delgado e sistema nervoso central.

38
Linfomas não Hodgkin Variam desde tumores de crescimento lento e comportamento indolente, até tumores muito agressivos: Indolentes - LLC, Linfoma folicular, Linfoma MALT Agressivos – Linfoma linfoblástico, Linfoma de grandes células difuso, Linfoma de Burkitt

39
Linfomas não Hodgkin na infância
Representam 10% dos tumores malignos das crianças Terceiro tumor mais frequente após as leucemias agudas e tumores cerebrais Subtipos agressivos, que frequentemente leucemizam e infiltram o SNC

40
Linfomas não Hodgkin nos adultos
Mais frequentes nos adultos do que nas crianças, aumentam a incidência com a idade Evolução agressiva ou arrastada de acordo com o subtipo histológico Tipos agressivos são curáveis com QT, enquanto os indolentes ficam sob controle, por até vários anos

42
Linfomas não Hodgkin Patogênese
Lesões genéticas, infecções virais e ativação de oncogens Translocações cromossômicas são as anormalidades mais frequentes Cada translocação costuma estar associada com um subtipo de linfoma

43
Linfomas não Hodgkin Patogênese
Solventes orgânicos, inseticidas, benzeno Radio e/ou quimioterapia prévias Imunodeficiências congênitas Imunodeficiências adquiridas: Transplantados e colagenoses

44
Linfomas não Hodgkin Patogênese
Infecções: HIV - associados a Linfomas não Hodgkin de origem B, bastante agressivos e com comprometimento extra nodal frequente Herpes vírus tipo 8 (HHV8) – Linfoma de cavidade. Associado à AIDS e caracterizado pela presença de derrames serosos (pleura, peritônio e pericárdio) sem adenomegalias.
 – Linfoma de cavidade. Associado à AIDS e caracterizado pela presença de derrames serosos (pleura, peritônio e pericárdio) sem adenomegalias.")
45
Linfomas não Hodgkin Patogênese
Infecções: Helicobacter pylori – causa a forma mais comum de linfoma extra nodal, o Linfoma MALT (Mucosa associated lymphoid tissue) do estômago HTLV1 – retrovírus associado à Leucemia/Linfoma T do adulto Hepatite C – Linfoma esplênico de zona marginal e outros linfomas B indolentes
 do estômago. HTLV1 – retrovírus associado à Leucemia/Linfoma T do adulto. Hepatite C – Linfoma esplênico de zona marginal e outros linfomas B indolentes.")
46
Linfomas não Hodgkin Patogênese
Infecções: Epstein Baar Virus associado com 100% dos casos de linfomas descritos por Burkitt, endêmico na África. Em países industrializados a expressão de EBV ocorre em percentual menor EBV presente em quase 100% dos linfomas B de grandes células e dos linfomas de Hodgkin em indivíduos portadores do HIV EBV presente em 100% dos casos de Linfomas de células T/NK nasofarígeo

47
Linfomas não Hodgkin Manifestações clínicas
Adenomegalias volumosas superficiais, cervicais, axilares, inguinais, ou em cadeias profundas,mediastino e abdominais Freqüentemente simétricas ou generalizadas, podem confluir e fusionar-se, formando grandes massas tumorais compressivas Adenomegalias com consistência de borracha e indolores De acordo com a velocidade de crescimento pode haver dor, sinais flogísticos e fistulização Compressão de estruturas pelas massas tumorais: Síndrome de veia cava superior, insuficiência respiratória e hidronefrose

48
Linfomas não Hodgkin Manifestações clínicas
Sintomas constitucionais: febre, sudorese noturna e emagrecimento Hepato e esplenomegalia podem ser precoces Podem ser acometidos sítios extra-nodais como pele, ossos, glândulas, SNC, TGI, pulmões Infiltração meníngea é comum nos linfomas leucemizados ou com infiltração da medula óssea, nas histologias agressivas

49
Linfomas não Hodgkin Manifestações clínicas
Infiltração da medula óssea causa citopenias: anemia, leucopenia e trombocitopenia Representação leucêmica no sangue periférico Derrames serosos por infiltração neoplásica ou por compressão podem ser precoces

51
Linfoma de Hodgkin Acomete ambos os sexos, com discreto predomínio do sexo masculino, raro nas crianças Distribuição etária com padrão bimodal – pico de incidência na 3a década de vida e um pico menor após os 50 anos Neoplasia maligna com origem sempre nos linfonodos, que são acometidos por contigüidade, ocorrendo disseminação hematogênica tardia, com invasão de medula óssea e fígado

52
Linfoma de Hodgkin Evolução clínica geralmente lenta e previsível, com alto percentual de cura com quimioterapia. Diagnóstico e sub-tipos histológicos baseiam-se na freqüência de: Célula de Reed Sternberg Ambiente de Hodgkin formado por linfócitos, plasmócitos, histiócitos, eosinófilos, neutrófilos e fibrose .

53
Linfoma de Hodgkin Fatores de risco
Predisposição genética e fatores familiares Infecção viral pelo EBV Infecção pelo HIV

54
Linfoma de Hodgkin Manifestações clínicas
Adenomegalias localizadas, geralmente unilaterais, mais freqüentemente em cadeias cervicais, principalmente à esquerda Consistência de borracha, indolores, evoluem por contigüidade e não se fusionam, nem fistulizam Massas mediastínicas são freqüentes Em estádios iniciais predominam as apresentações supra-diafragmáticas

55
Linfoma de Hodgkin Manifestações clínicas
Sintomas constitucionais são freqüentes e podem simular doenças infecciosas: Febre, sudorese noturna e perda de peso > 10% peso em 6 meses Pode ocorrer regressão parcial ou total, temporária das adenomegalias Pode evoluir com eosinofilia e linfopenia. Quando infiltra a medula óssea pode causar pancitopenia

56
Linfoma de Hodgkin Manifestações clínicas
Infiltração do baço ocorre antes da infiltração hepática Infiltração da medula óssea, do fígado e os derrames serosos são raros em fases iniciais e frequentes na doença avançada Não se representa no sangue periférico Não causa doença exclusivamente extra-nodal Infecções por comprometimento da imunidade celular como Tuberculose, toxoplasmose, Pneumocistose, criptococose, histoplasmose, estrongiloidíase

57
Diagnóstico dos Linfomas
Comprovação histopatológica através de biópsia ganglionar ou de outro sítio envolvido, como por exemplo medula óssea, pele, estômago Exame citológico por PAAF não permite estudar a arquitetura do gânglio e dificulta a classificação do linfoma. Ajuda na avaliação da recidiva

58
Diagnóstico dos Linfomas
Retirar sempre que possível o maior gânglio, INTEIRO, com sua cápsula Fazer o “inprint”, para exame citológico, após seccionar o gânglio no maior sentido. Deixar secar as lâminas e corar pelo Giemsa Logo após seccioná-lo, colocar o linfonodo no fixador formol tamponado e enviar ao laboratório

59
Diagnóstico dos Linfomas
Se houver adenomegalia generalizada, dar preferência para a biópsia nesta ordem gânglios cervicais gânglios axilares gânglios inguinais Biopsiar gânglios intra torácicos ou intra abdominais se não houver adenomegalias periféricas

60
Diagnóstico dos Linfomas
Aspectos importantes Envio dos dados clínicos completos ao patologista Avaliação conjunta dos casos pelo patologista especializado com o hematologista Complementação do diagnóstico morfológico com a Imunohistoquímica

61
Estadiamento dos Linfomas
Procedimento obrigatório para determinar o grau de extensão da doença Necessário para escolha do plano terapêutico O estadiamento dos Linfomas baseia-se em: Localização dos linfonodos Presença ou ausência de visceromegalias Doença extra nodal Presença ou ausência de sintomas constitucionais Exames complementares

62
Estadiamento dos Linfomas
Anamnese e exame físico Hemograma, plaquetas e VHS Bioquímica Sorologias Biópsia e aspirado de medula óssea Endoscopia digestiva, se houver clínica Biópsias de lesões suspeitas Exame citológico dos derrames Tomografia computadorizada

63
Evolução, prognóstico e tratamento Linfomas não Hodgkin
Os Linfomas de comportamento indolente raramente são diagnosticados em fases iniciais Caso sejam localizados, podem ser curados com Radioterapia Quando disseminados,respondem bem à quimioterapia, mas não são curáveis, recidivando várias vezes ao longo de anos e podendo sofrer transformação para formas histológicas mais agressivas

64
Evolução, prognóstico e tratamento Linfomas não Hodgkin
Os Linfomas agressivos são curáveis com QT que pode ser associada à RT e/ou AcMo anti CD20 Muitos casos são resistentes e recidivam precocemente, com alta mortalidade O Transplante de medula óssea, geralmente autólogo, é indicado para os Linfomas agressivos sensíveis à QT, em recidiva.

65
Evolução, prognóstico e tratamento Linfomas de Hodgkin
Evolução lenta, acometendo novas regiões nodais por contigüidade e disseminação hematogênica tardia Alto percentual de curas com radioterapia e/ou quimioterapia, mesmo nos estádios avançados Pode recidivar anos mais tarde, permanecendo uma doença curável com associação de QT de 2a linha e TMO autólogo A associação de QT e RT aumenta a chance de neoplasias secundárias hematológicas como as LMA e os linfomas não Hodgkin.

66
Adenomegalias cervicais

67
Adenomegalias cervicais

68
Adenomegalias axilares

69
Apresentação extra nodal

71
Célula de Reed Sternberg

72
Linfoma de Hodgkin

73
Micose fungóide

74
Linfoma T periférico

75
Linfoma T/NK

76
Apresentação extranodal

77
Linfoma de Burkitt

78
GAMOPATIAS MONOCLONAIS

79
Gamopatias Monoclonais
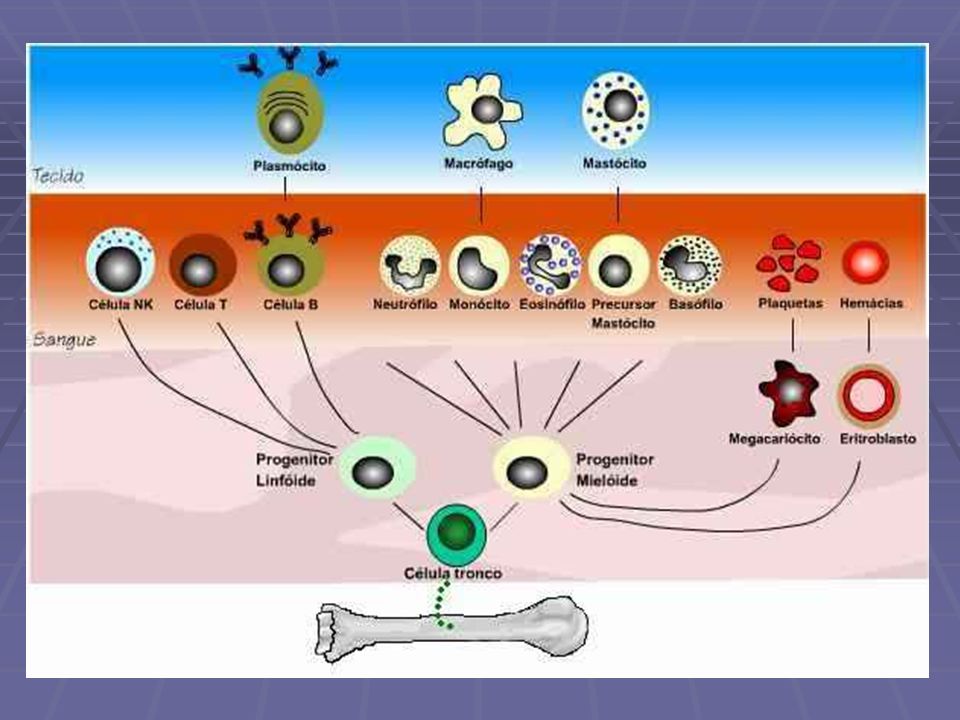
Distúrbios Leucocitários Leucopênicos Proliferativos Reativos Neoplásicos Linfóides Leucemias Linfomas Plasmocitomas Mielóides

80
Gamopatias Monoclonais
Definição: grupo de doenças caracterizadas pela existência de um clone neoplásico de plasmócitos ou linfócitos secretores de imunoglobulina homogênea ou seus componentes, que é reconhecida como um pico característico na eletroforese de proteínas séricas ou urinárias e que podem resultar na deposição de cadeias leves ou pesadas em vários tecidos. Arch Pathol Lab Med 1999; 123: / JBN 2003; 25(4):224-7
:")
81
Gamopatias Monoclonais
Estímulos Ambientais Predisposição Genética Patogenia Expressão de oncogenes Inibição de genes supressores Anormalidade Citogenética Trissomias Monossomia Translocações Hipodiploidias Deleções

82
Gamopatias Monoclonais

83
Gamopatias Monoclonais
Epidemiologia Arch Pathol Lab Med 1999; 123:

84
Mieloma Múltiplo Definição: Neoplasia plasmocitária com comprometimento multilocalisado do esqueleto. Histórico: Descrições de Willian Macintyre e Henry Bence Jones em 1845 Epidemiologia paises industrializados: 4/ países subdesenvolvidos: <1/ brancos x negros : 4/ x 10/ Idade > 55 anos homens x mulheres: 3:2 1% das mortes por câncer

85
Mieloma Múltiplo Sintomas Clínicos e resultados laboratoriais
dor óssea (70%) radiografia com lesões líticas mal estar geral e queixas indefinidas perda de peso infecções recorrentes Anemia VHS aumentado sintomas associados à hipercalcemia cansaço, sede, náuseas, constipação, etc. sintomas associados à hiperviscosidade plasmática hemorragias, sonolência, sintomas neurológicos isqêmicos, etc. CRAB: Hipercalcemia, Insuficiência renal, Anemia, Lesão óssea
 radiografia com lesões líticas. mal estar geral e queixas indefinidas. perda de peso. infecções recorrentes. Anemia. VHS aumentado. sintomas associados à hipercalcemia. cansaço, sede, náuseas, constipação, etc. sintomas associados à hiperviscosidade plasmática. hemorragias, sonolência, sintomas neurológicos isqêmicos, etc. CRAB: Hipercalcemia, Insuficiência renal, Anemia, Lesão óssea.")
86
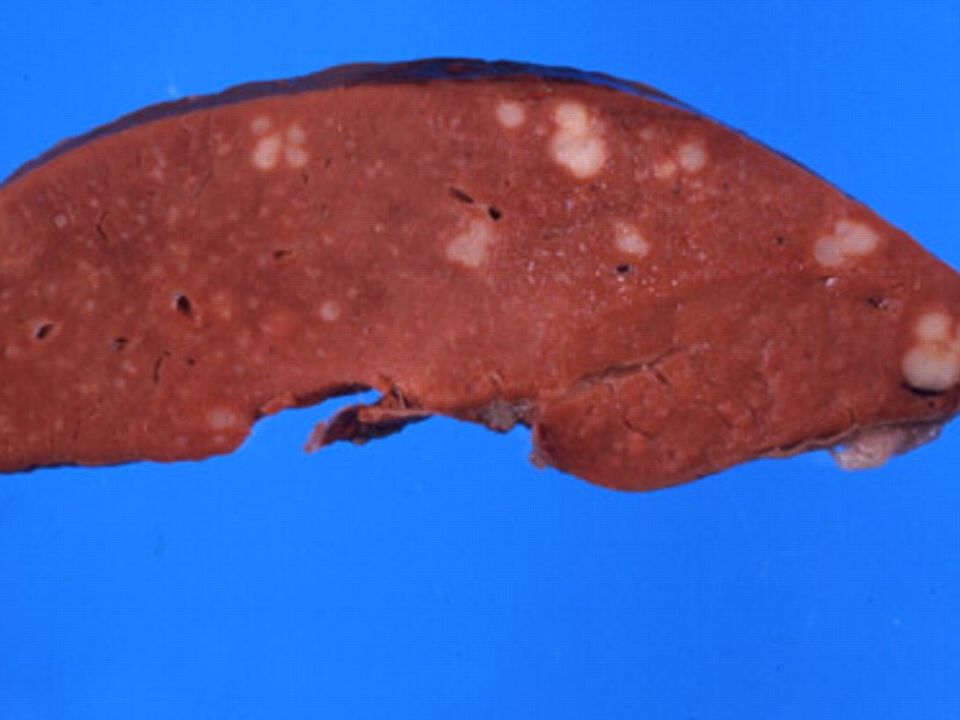
Mieloma Multiplo: lesões líticas e fraturas patológicas

87
Mieloma Múltiplo Distribuição das lesões ósseas coluna vertebral: 66%
costelas: 44% crânio: 41% pelve: 28% femur: 24% clavícula: 10% escápula: 10%

88
Mieloma Múltiplo Diagnóstico clínico-patológico
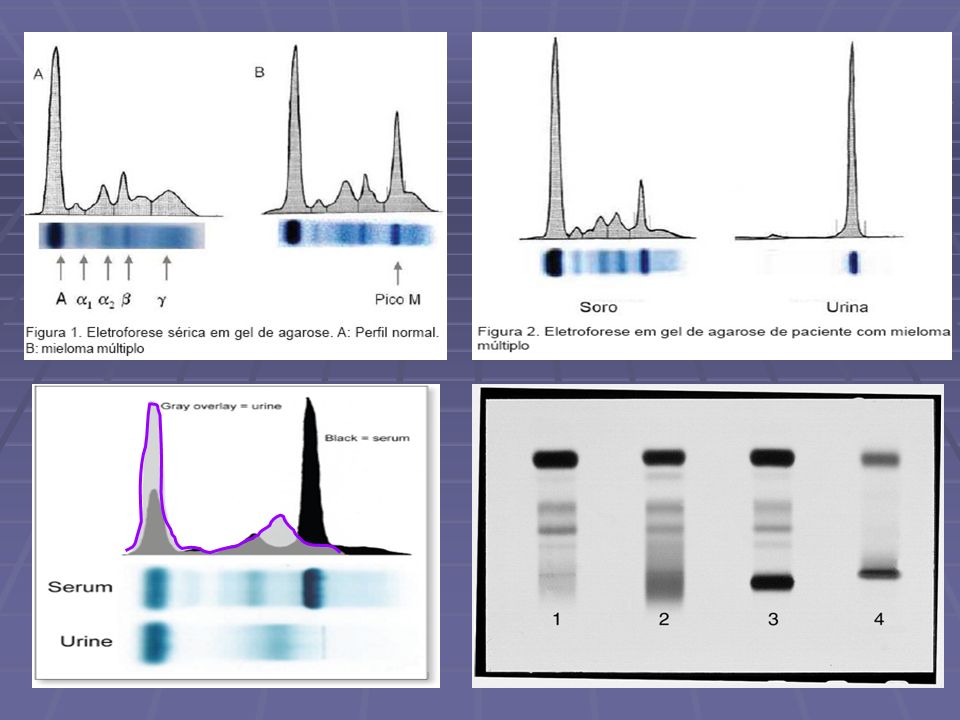
Exames Radiologicos radiografia por raios X ressonância magnética Eletroforese de proteínas séricas detecção de proteína monoclonal (pico monoclonal ou banda M) estimativa da concentração da imunoglobulina monoclonal Eletroforese de proteínas urinárias detecção e quantificação da proteína de Bence Jones Imunoeletroforese ou imunofixação qualificação e quantificação das imunoglobulinas monoclonais e/ou seus componentes Doseamento de imunoglobulinas por nefelometria Biópsia de medula óssea
 estimativa da concentração da imunoglobulina monoclonal. Eletroforese de proteínas urinárias. detecção e quantificação da proteína de Bence Jones. Imunoeletroforese ou imunofixação. qualificação e quantificação das imunoglobulinas monoclonais e/ou seus componentes. Doseamento de imunoglobulinas por nefelometria. Biópsia de medula óssea.")
90
Gamopatias Monoclonais
Tipos de imunoglobulina monoclonal % IgG 52 Mais comum IgA 21 Doença extramedular IgD 2 Leucemização IgE 0,01 IgM 12 Macroglobulinemia de Waldenstrom Dois tipos (biclonal) <1 Apenas cadeias pesada Ausência de proteina M 1 Mieloma não secretor Apenas PBJ 11
 <1. Apenas cadeias pesada. Ausência de proteina M. 1. Mieloma não secretor. Apenas PBJ. 11.")
91
Gamopatias Monoclonais
Mieloma Solitário plasmocitoma medular ou extramedular Imunoglobulina monoclonal <2,5 g/dL e PBJ<500 mg/dL 3-5% das neoplasias de plasmócitos estágio inicial do Mieloma Múltiplo (?) Síndrome POEMS (Peripheral neuropathy, Organomegaly, Endocrine deficiency, Monoclonal gammopathy and Skin pigmentation) síndrome rara com neuropatia periférica sobrevida de 5 anos
 Síndrome POEMS (Peripheral neuropathy, Organomegaly, Endocrine deficiency, Monoclonal gammopathy and Skin pigmentation) síndrome rara com neuropatia periférica. sobrevida de 5 anos.")
92
Gamopatias Monoclonais
Macroglobulinemia de Waldenstron secreção de IgM monoclonal síndrome da hiperviscosidade fluxo sanguíneo dificultado anemia hemorragia neuropatia amiloidose

93
Gamopatias Monoclonais
Amiloidose deposição de proteína fibrilar formada por cadeias λ nefrose cardiomiopatia DPOC distúrbios absortivos neuropatia periférica púrpura síndrome do túnel do carpo Doença de Deposição de Imunoglobulinas Deposição de cristais de imunoglobulina monoclonal ou de seus componentes síndrome nefrótica e insuficiência renal

94
Gamopatias Monoclonais
Doença de Deposição de Imunoglobulinas Deposição de cristais de imunoglobulina monoclonal ou de seus componentes Nefropatia por cadeias leves Nefropatia por cadeias pesadas Nefropatias por cadeias leves e pesadas

95
Gamopatias Monoclonais
Gamopatia Monoclonal de Significado Indeterminado Crônica 1% dos indivíduos >50a e 3% dos indivíduos >70a 1,5% evoluem para MM, MW ou Amiloidose Sobrevida de 12 anos Critérios para GMSI Imunoglobulina monoclonal <2,5g/dL PBJ <50 mg/dL Plasmócitos medulares <10% Sem lesões ósseas Sem supressão imunológica Transitória Gamopatia Monoclonal Associado ao Transplante

96
HISTIOCITOSE X Histiocitose de Células de Langerhans.
Proliferação de células do sistema monocítico-macrofágico e dendríticas. Pode ser devido a estimulação imunológica. Natureza não neoplásica?

97
HISTIOCITOSE X Pico entre 1-4 anos; 2:1 em meninos.
Formas clínicas: 1) Sd Letterer Siwe – Trombocitopenia, anemia, linfadenopatia. Manifestações bucais: ulcerações, hiperplasia gengival, destruição óssea alveolar; 2) Hand-Schuller-Christian – crianças e adultos jovens, Adenopatia é o sinal inicial. Lesão lítica dos ossos, dor óssea, fraturas patológicas. Acompanhada de diabetes insípido e exoftalmia. Hepatoesplenomegalia, lesões pulmonares, infecções recorrentes, febre, erupções difusas.
 Sd Letterer Siwe – Trombocitopenia, anemia, linfadenopatia. Manifestações bucais: ulcerações, hiperplasia gengival, destruição óssea alveolar; 2) Hand-Schuller-Christian – crianças e adultos jovens, Adenopatia é o sinal inicial. Lesão lítica dos ossos, dor óssea, fraturas patológicas. Acompanhada de diabetes insípido e exoftalmia. Hepatoesplenomegalia, lesões pulmonares, infecções recorrentes, febre, erupções difusas.")
98
2) Hand-Schuller-Christian – Manifestações bucais – sinal precoce: gengivite, halitose ulceras, perda do osso alveolar semelhante a doença periodontal; 3) Granuloma Eosinofilico – região posterior da mandíbula – benigno; Diagnostico Diferencial – Cisto, doença periodontal avançada, Linfoma de burkitt; Osteomielite; sarcomas e milema múltiplos;
 Granuloma Eosinofilico – região posterior da mandíbula – benigno; Diagnostico Diferencial – Cisto, doença periodontal avançada, Linfoma de burkitt; Osteomielite; sarcomas e milema múltiplos;")
99
HISTIOCITOSE X ANATOMOPATOLÓGICO: infiltrado inflamatório.
Presença de marcadores imuno-histoquímicos e aumento de citocinas. O diagnóstico definitivo é feito quando lesões suspeitas apresentam grânulos de Birbeck na microscopia eletrônica e demonstrar antígeno T6 nas suas superfícies. Essas características auxiliam a diferenciar os pacientes com histiocitoses de células de Langerhans daqueles que apresentam desordens histiocíticas não-Langerhans. Pancitopenia → mielograma e/ou biópsia de medula óssea. do VHS, disfibrinogenemia, hipoalbuminemia e ascite.

100
" O que mais preocupa não é nem o grito dos violentos, dos
corruptos, dos desonestos, dos sem caráter, dos sem ética. O que mais preocupa é o silêncio dos bons.“ Martin Luther King Obrigado!!!! Dúvidas:

Apresentações semelhantes















>")





.>")



