Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Talassemias e Hemoglobinopatias
.

2
Composição das hemoglobinas
Tipo de Hemoglobina Cadeias globina Adulto Recém-nascido A (=A1) α2β2 96 – 97% 20% A2 α2δ2 2,5% 0,5% F (Fetal) α2γ2 < 1% 80% Gower 1 ζ2ε2 - variável-embrião Portland ζ2γ2 Gower 2 α2ε2 H β4 α-Talassemia (vida adulta) Bart’s γ4 α-Talassemia (vida fetal) + GRUPO HEME
 α2β2. 96 – 97% 20% A2. α2δ2. 2,5% 0,5% F (Fetal) α2γ2. < 1% 80% Gower 1. ζ2ε2. - variável-embrião. Portland. ζ2γ2. Gower 2. α2ε2. H. β4. α-Talassemia. (vida adulta) Bart’s. γ4. α-Talassemia (vida fetal) + GRUPO HEME.")
3
4 semanas: Hb Gower-1 (ζ2ε2) > 25ª semana: Hb Fetal (α2γ2) :90-100%
Hb A(α2β2): 5-10% Hb A2(α2δ2): traços 12 – 25ªsemana: Hb Gower-1(ζ2ε2): 20-40% Hb Portland: (ζ2γ2) 5-20% Hb Gower-2 (α2ε2): 10-20% Após Nascimento: Hb A: 96-98% Hb A2: 2-3,5% Hb F: 0-1,0%
: 5-10% Hb A2(α2δ2): traços. 12 – 25ªsemana: Hb Gower-1(ζ2ε2): 20-40% Hb Portland: (ζ2γ2) 5-20% Hb Gower-2 (α2ε2): 10-20% Após Nascimento: Hb A: 96-98% Hb A2: 2-3,5% Hb F: 0-1,0%")
4
Talassemias Grupo heterogêneo de doenças genéticas resultantes da diminuição da velocidade de síntese de cadeias α e β. São 44 tipos variantes de talassemias identificadas Classificação: α β 0 ausência ou diminuição acentuada na síntese da globina α β + síntese insuficiente da globina

6
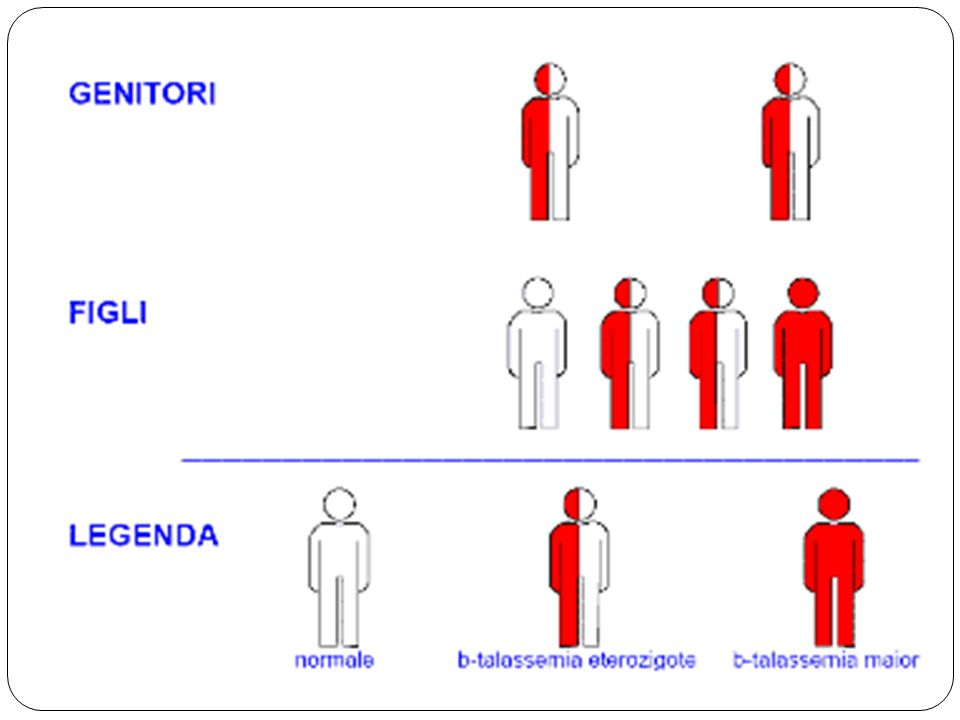
Clínica Talassemia Hidropsia fetal Talassemia Maior
β0-talassemia ou outras combinações do traço β-talassêmico Talassemia Intermédia vide quadro 1 Talassemia menor Traço β0-talassemico Traço β+ -talassemico Persistência hereditária de hemoglobina fetal Traço δβ-talassêmico Traço α0-talassêmico Traço α+-talassêmico Tabela 1

7
Talassemia intermédia – quadro 1
β-talassemia homozigótica β+- talassemia homozigótica leve Herança concomitante de α-talassemia Aumento da capacidade de produzir hemoglobina fetal (produção de cadeia γ) β-talassemia heterozigótica Herança concomitante de genes adicionais de α-globina (ααα/αα, ααα/ααα) Traço β-talassêmico dominante δβ-talassemia e persistência hereditária de hemoglobina fetal δβ -talassemia homozigótica δβ-talassemia hereozigótica/β-talassemia Hb Lepore homozigótica (alguns casos) Hemoglobinopatia H
 β-talassemia heterozigótica. Herança concomitante de genes adicionais de α-globina (ααα/αα, ααα/ααα) Traço β-talassêmico dominante. δβ-talassemia e persistência hereditária de hemoglobina fetal. δβ -talassemia homozigótica. δβ-talassemia hereozigótica/β-talassemia. Hb Lepore homozigótica (alguns casos) Hemoglobinopatia H.")
8
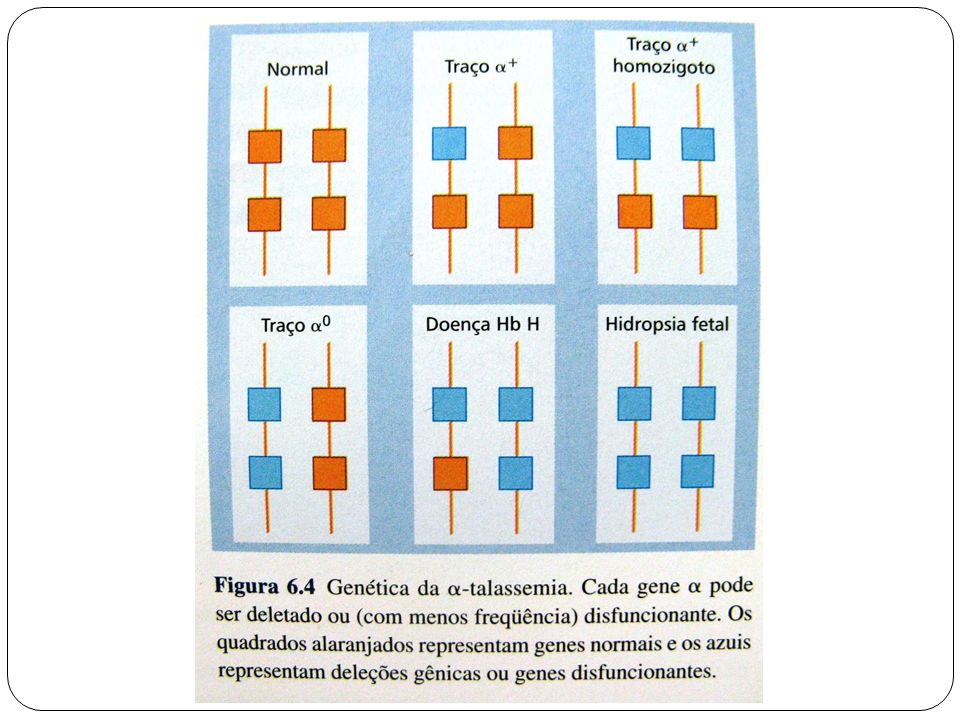
Alfa-Talassemia Anemia microcítica hipocrômica
Presença de Hb H ou Hb Bart´s Presença de precipitado intra-eritrocitário de Hb H Hemoglobina A2 normal Ferritina normal Biologia molecular

10
Alfa-Talassemia Exames obrigatórios
Hemograma Reticulócitos Eletroforese de Hb - A2 normal ou diminuída (3 genes alterados/deletados) Pesquisa de Hb H Ferritina Pesquisa de Alfa-Talassemia por Biologia Molecular
 Pesquisa de Hb H. Ferritina. Pesquisa de Alfa-Talassemia por Biologia Molecular.")
11
HEMOGLOBINOPATIA H (α-talassemia)
")
12
Caso Clínico: S.R. – branca – 40 anos - feminino
Ht: V.C.M: 72 Hb: 10, H.C.M: 23 Hct: 33, C.H.C.M: 32 Ferritina: 280 (15 – 80) Eletroforese de Hb: A (AA)- 96,4% Hb A2: 2,8 % Hb F: 0,8% Pesquisa de Hb H: positiva
 Eletroforese de Hb: A (AA)- 96,4% Hb A2: 2,8 % Hb F: 0,8% Pesquisa de Hb H: positiva.")
14
Beta-talassemia classificação clínica
Beta Talassemia Silenciosa Beta Talassemia Menor Beta Talassemia Intermédia Beta Talassemia Maior

15
Beta Talassemia: fisiopatologia
α α / β Precipitação da cadeia α Pontilhado basófilo nas hemácias

16
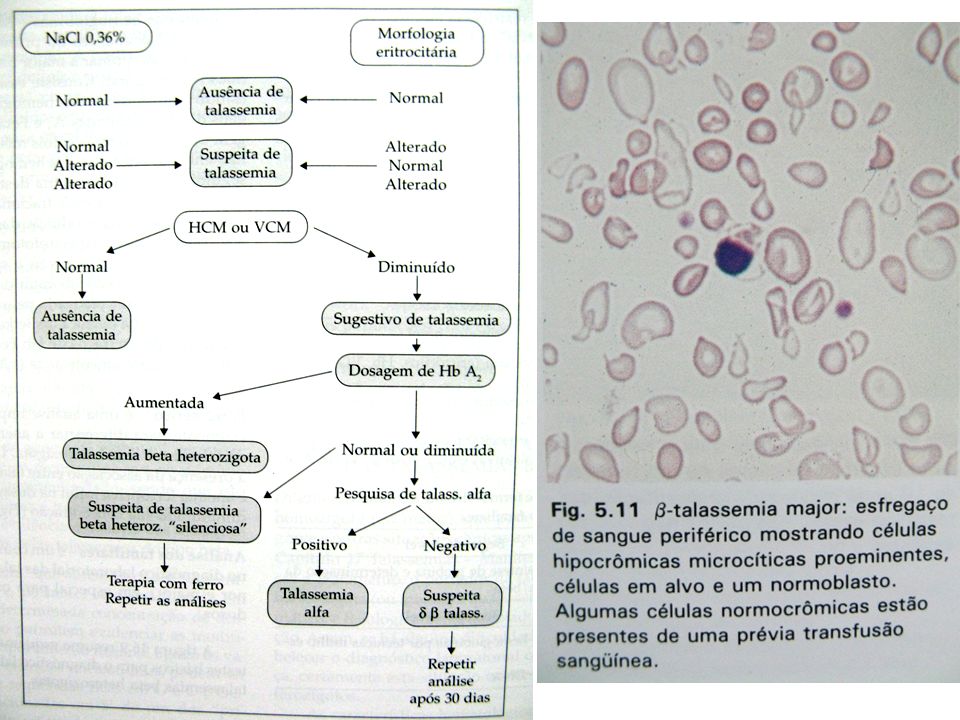
Beta Talassemia Menor Quadro Laboratorial
Anemia discreta ou ausente Morfologia: microcitose, hipocromia, hemácias elevadas (>5,5M/μL), poiquilocitose, ovalocitose, pontilhado basófilo VCM 61 a 73 fL Hb A2 aumentada (4 a 7%) Hb Fetal normal ou pouco aumentada Ferritina normal
, poiquilocitose, ovalocitose, pontilhado basófilo. VCM 61 a 73 fL. Hb A2 aumentada (4 a 7%) Hb Fetal normal ou pouco aumentada. Ferritina normal.")
17
Caso clínico L.D.B., mulher, 62 anos; sem informações clínicas.
HEM = 5,57M/µL HGB = 11,90 g/dL HCT = 36,4 % VCM = 65,35 fL HCM = 21,36 pg CHCM= 32,69 % RDW = 18,30 Morfologia eritrocitária: Anisocitose moderada com Microcitose; Poiquilocitose Moderada com Hemácias em Alvo, eliptócitos, ovalócitos e dacriócitos; Policromasia (++/5) e Pontilhados Basófilos (++/5).
 e Pontilhados Basófilos (++/5).")
18
Caso clínico Y.M.G.S., feminino, 5 anos. RBC = 5,8 M/µL
HGB = 12,5 g/dL HCT = 39,3% VCM = 67,76 fL HCM = 21,55 pg CHCM= 31,8% RDW = 14,6 Morfologia Eritrocitária: Microcitose; Poiquilocitose Leve com eliptócitos, hemácias em alvo e em lágrima.

19
Mulher de 35 anos;

20
Mulher de 35 anos;

21
Beta Talassemia Maior Clínica
Anemia grave – 3 a 4 meses após o nascimento Hepatoesplenomegalia Hematopoese extramedular Expansão dos ossos causada pela intensa hiperplasia eritróide da M.O.

22
Beta Talassemia maior Diagnóstico laboratorial
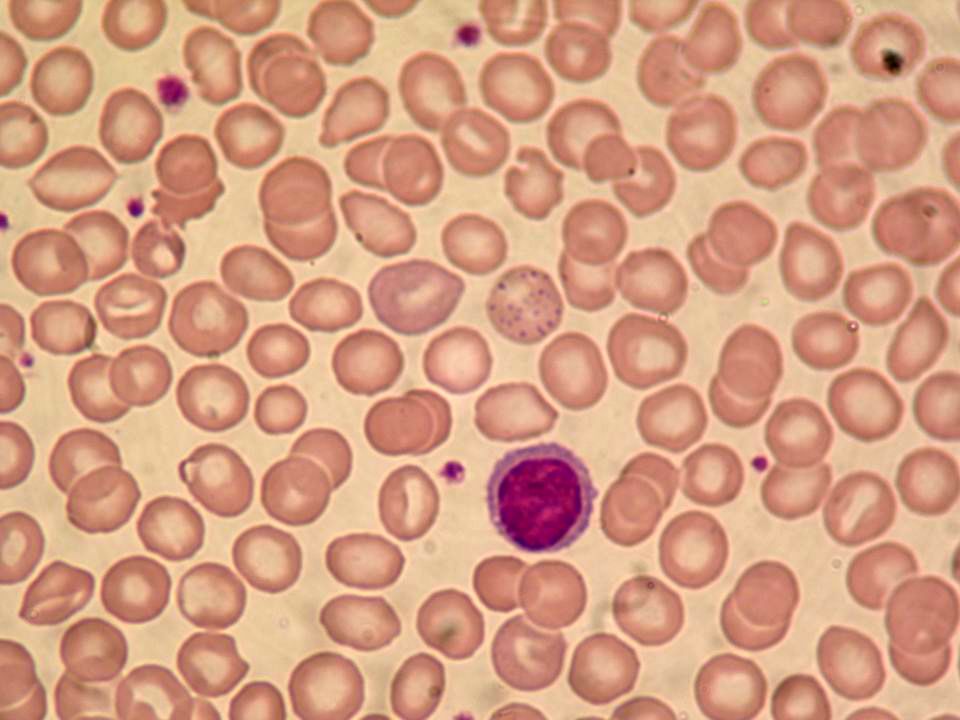
Anemia microcítica (extrema) e hipocrômica Reticulocitose e presença de eritroblastos circulantes Células em alvo Pontilhado basófilo Eletroforese: ausência ou acentuada diminuição de Hb A (α2β2); Hb F (α2γ2) perfaz quase toda Hb circulante; Hb A2 (α2δ2) normal ou levemente alta (geralmente > 3,5%) Biologia molecular – análise de DNA
 e hipocrômica. Reticulocitose e presença de eritroblastos circulantes. Células em alvo. Pontilhado basófilo. Eletroforese: ausência ou acentuada diminuição de Hb A (α2β2); Hb F (α2γ2) perfaz quase toda Hb circulante; Hb A2 (α2δ2) normal ou levemente alta (geralmente > 3,5%) Biologia molecular – análise de DNA.")
25
Talassemia Intermédia
Anemia moderada (Hb g/dL) δβ-Talassemia: 5 – 20% Hb F Hemoglobina de Lepore: ausência de síntese de cadeias δ e β Diagnóstico: Eletroforese de hemoglobina
 δβ-Talassemia: 5 – 20% Hb F. Hemoglobina de Lepore: ausência de síntese de cadeias δ e β. Diagnóstico: Eletroforese de hemoglobina.")
26
Beta Talassemia: Portador Silencioso
Filho com Talassemia maior e somente um dos pais tem talassemia menor Ausência de anemia Microcitose discreta Hb A2: normal Biologia molecular: discreta redução na síntese da cadeia beta

28
Tipo de talassemia β+ β + Mediterrâneo β+ Africano Sinônimo clínico %
HOMOZIGOTO HETEROZIGOTO Tipo de talassemia Sinônimo clínico Anemia % Hb F % HbA2 β+ Cooley, T. maior ++++ ~ 90 N ou ↑ T. Menor + / - Pouco elevada em 50% dos casos > 3,5 β + Mediterrâneo Cooley +++ 20 – 80 β+ Africano Talassemia intermédia ++ 20 – 40 Pouco elevada ou N

29
Tipo de talassemia β+ Silencioso β0 ou β+ com Hb A2 normal
HOMOZIGOTO HETEROZIGOTO Tipo de talassemia Sinônimo clínico Anemia % Hb F % HbA2 β+ Silencioso Talassemia intermédia + ou ++ 10 – 30 ↑ - N > 3,5 β0 ou β+ com Hb A2 normal Provável Talassemia intermédia ++ ou +++ 20 – 80 T.Menor + / - 2,0 – 3,5 + discreta; ++ moderada; +++ grave; muito grave; - ausente; N – normal; ↑ - aumentada

30
Indivíduo normal

31
Anemia de Células Falciformes
Defeito genético causado pela substituição de um aa na posição 6 da cadeia β: ácido glutâmico por valina Hb S (Hb α2β2s) insolúvel na água quando exposta a baixas tensões de oxigênio, ocorre polimerização em fibras longas (formada por 7 fios duplos enrolados com ligações cruzadas) Síndrome falcêmica com homozigose (Hb SS). (S de sicle = foicinha)
 insolúvel na água quando exposta a baixas tensões de oxigênio, ocorre polimerização em fibras longas (formada por 7 fios duplos enrolados com ligações cruzadas) Síndrome falcêmica com homozigose (Hb SS). (S de sicle = foicinha)")
32
Fonte: http://www. scielo. br/scielo. php

33
Anemia Falciforme – Manifestações clínicas:
Anemia grave: crises (a severidade clínica varia de paciente para paciente); Crises vasoclusivas dolorosas; Crises de sequestro visceral; Crises aplásticas; Crises hemolíticas: 1/3 intravascular, 2/3 extravascular
; Crises vasoclusivas dolorosas; Crises de sequestro visceral; Crises aplásticas; Crises hemolíticas: 1/3 intravascular, 2/3 extravascular.")
34
Anemia Falciforme – Manifestações clínicas:
Outras: úlceras nas extremidades das pernas; esplenomegalia em lactentes e início da infância; hipertensão pulmonar; retinopatia proliferativa; lesão crônica do fígado (com microinfartos); infartos renais; osteomielite (geralmente por Salmonella sp).
; infartos renais; osteomielite (geralmente por Salmonella sp).")
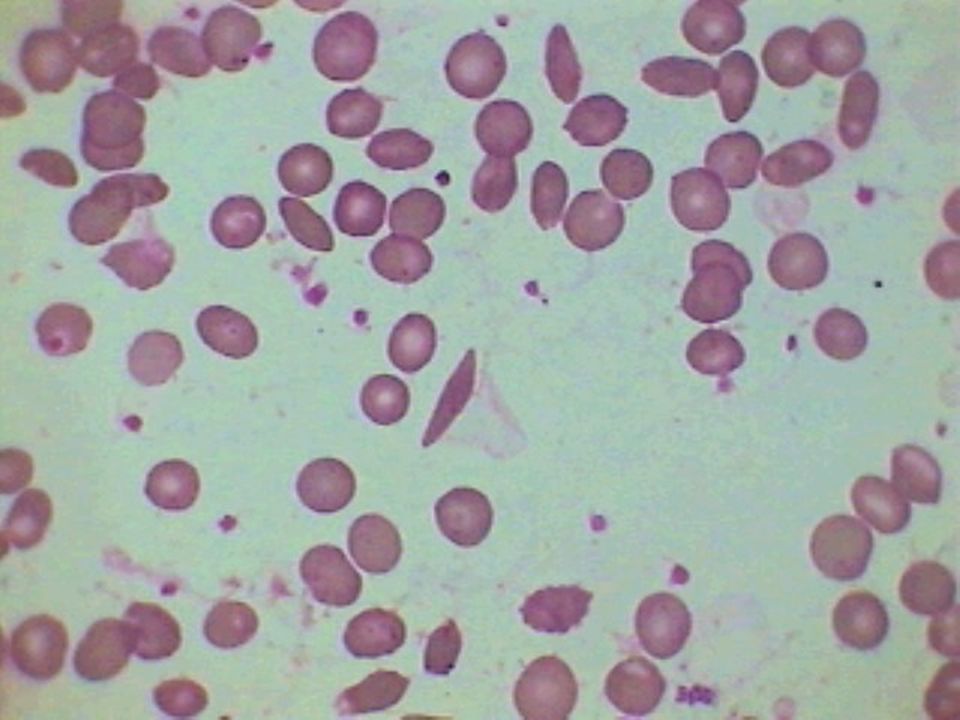
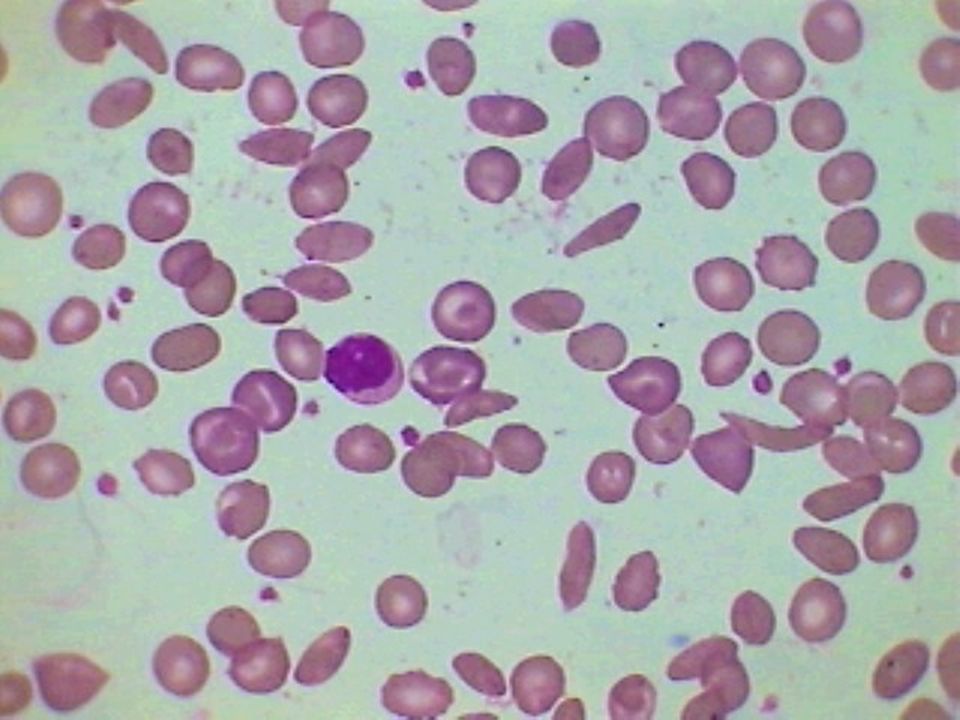
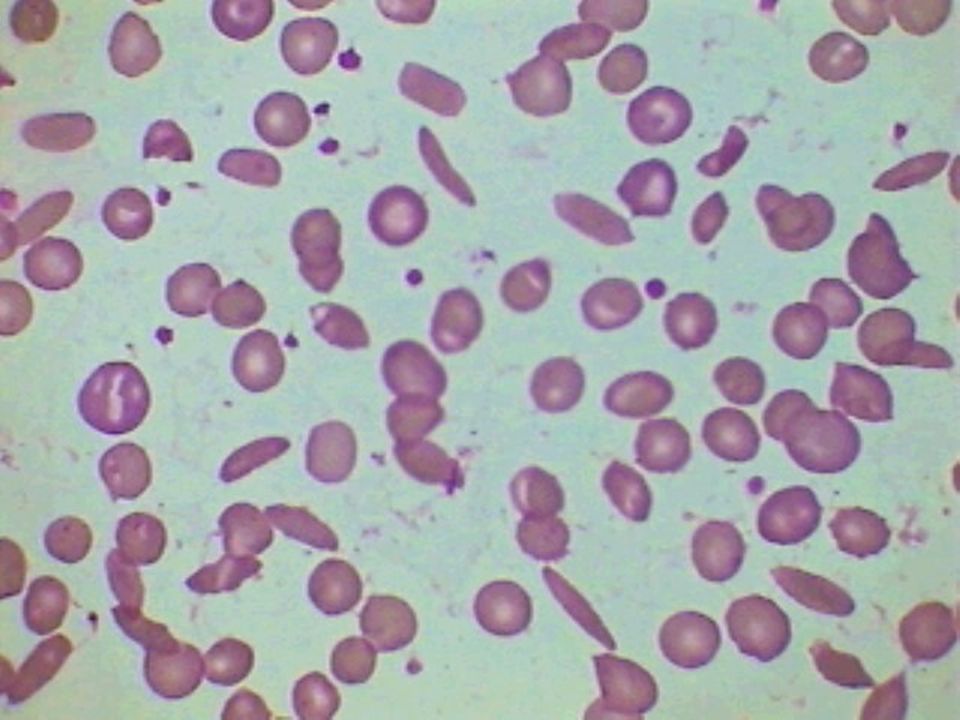
36
Anemia Falciforme – Achados Laboratoriais:
Hemoglobina entre 6 a 9 g/dL Morfologia eritrocitária: anisocitose acentuada, Células falciformes e células em alvo, corpúsculos de Howell-Jolly, policromatocitose (reticulocitose) e eritroblastos circulantes. Eletroforese de hemoglobina: SS Hb A não é detectada, Hb F de 5 a 15%
 e eritroblastos circulantes. Eletroforese de hemoglobina: SS Hb A não é detectada, Hb F de 5 a 15%")
37
ELETROFORESE EM PH ALCALINO

38
Caso clínico Hemácias: 2,42 M/μL Hemoglobina:7,9 g/dL
Hematócrito:22.0 % VCM: 90.0 fL HCM: 32.6 pg CHCM: 35.9 % RDW: 20.3 LEUCÓCITOS: /μL Plaquetas: /μL

42
CASO – TESTE DE FALCIZAÇÃO: POSITIVO

43
CASO MORFOLOGIA ERITROCITÁRIA:
Anisopoiquilocitose com hemácias falciformes, hemácias em alvo e policromasia

44
Traço de Células Falciformes
Condição benigna sem anemia; Morfologia eritrocítica de aspecto normal Hematúria é o sintoma mais comum Hb S 25 a 45% Cuidados especiais: anestesia, gravidez e subida a grandes altitudes

45
Combinação de Hb S com outros defeitos genéticos da Hemoglobina
Mais comuns: Hb S/β-talassemia VCM e HCM mais baixos que na anemia falciforme Hb S/C tendência a tromboses e embolia pulmonar

46
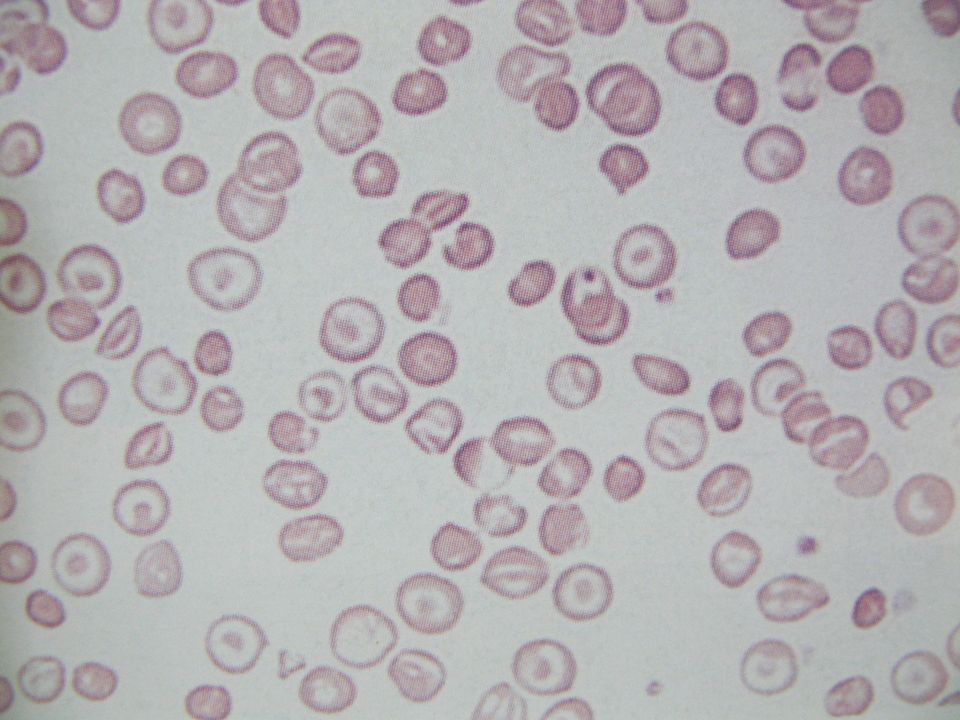
Hemoglobinopatia C Substituição na posição 6 da cadeia β do ácido glutâmico por lisina Tendência a formar cristais romboidais nos eritrócitos Homozigose (C/C): - esplenomegalia com anemia hemolítica revelando células em alvo (elevada porcentagem no esfregaço), microesferócitos e eritrócitos com forma romboidal Heterozigose (A/C): distensão sanguínea com eritrócitos em alvo, não revelando crises hemolíticas.
: - esplenomegalia com anemia hemolítica revelando células em alvo (elevada porcentagem no esfregaço), microesferócitos e eritrócitos com forma romboidal. Heterozigose (A/C): distensão sanguínea com eritrócitos em alvo, não revelando crises hemolíticas.")
48
Hemoglobinopatia D Grupo de variantes com mesma mobilidade eletroforética Heterozigotos não apresentam anomalias hematológicas e os homozigotos têm anemia hemolítica leve.

49
Hemoglobinopatia E Variante de hemoglobina mais comum no sudeste da Ásia. β26 Lis Glu. Homozigoto: anemia microcítica hipocrômica leve Hemoglobinopatia E/β0-talassemia: manifestações semelhantes à talassemia maior (clínica e hematologicamente)
")
Apresentações semelhantes



>")


















